
{kind=link}
| Author | Affiliation |
|---|---|
| Edward J. LoCascio, MD | Louisiana State University Health Sciences Center, Shreveport, LA |
| Simon A. Mahler, MD | Louisiana State University Health Sciences Center, Shreveport, LA |
| Thomas C. Arnold, MD | Louisiana State University Health Sciences Center, Shreveport, LA |
ABSTRACT
Emergency physicians (EP) frequently encounter angioedema involving the lips and tongue. However, angioedema from Angiotensin Converting Enzyme inhibitors or hereditary angioedema (HAE) can present with gastrointestinal symptoms due to bowel wall involvement. EPs should begin to consider this clinical entity as a potential cause for abdominal pain and associated gastrointestinal symptoms given the common use of medications that can precipitate angioedema. We report a case of a 34-year-old woman who presented with abdominal cramping, vomiting and diarrhea due to an acute exacerbation of HAE.
INTRODUCTION
Intestinal angioedema is less commonly encountered by emergency physicians (EP) than angioedema of the lips and tongue and therefore may be unrecognized. Prompt detection and treatment of this rare disease can significantly improve patient outcome by minimizing morbidity from misdiagnosis or unnecessary operative interventions. EPs should be aware that hereditary angioedema (HAE) and medications, such as Angiotension Converting Enzyme (ACE) inhibitors, can cause intestinal angioedema and therefore may present with gastrointestinal complaints. In this case report, we will review the features that may help differentiate intestinal angioedema from other, more common, etiologies of abdominal pain with gastrointestinal symptoms. We also provide a review of the literature including therapy, which remains controversial in the emergency department (ED) setting.
A review of emergency medicine (EM) literature revealed no previously reported cases of isolated intestinal angioedema caused by HAE. A few such cases have been described in radiology and gastroenterology literature spanning several decades.1–9 However, a case of ACE-inhibitor associated intestinal angioedema was recently described in the EM literature.10
CASE REPORT
A 34-year-old African-American female who denied any prior medical history presented to the ED complaining of a three-day history of diffuse, constant abdominal “cramping” with associated nausea, vomiting and blood-streaked diarrhea. She received two liters of crystalloid and 25 mg of promethazine intravenously in the triage area and reported resolution of her nausea at the time of the initial interview by the EP. She described four similar episodes in the preceding year, for which she had been seen in the ED, treated symptomatically with intravenous fluids and promethazine and discharged with a diagnosis of gastroenteritis. She did note, however, that no other relatives or personal contacts were suffering from these symptoms.
Physical examination revealed vital signs consisting of an oral temperature of 36.7°C (98.0°F); blood pressure 137/106 mm Hg; heart rate 114 beats/min; and a respiratory rate of 18 breaths/min. She appeared in no apparent distress, was alert, awake, and oriented. Pertinent physical examination findings included the presence of normoactive bowel sounds and tenderness to palpation in the bilateral lower abdominal quadrants. She exhibited some voluntary guarding but had no rebound tenderness, percussive tenderness, or involuntary guarding. There were no remarkable cutaneous lesions visualized, nor were any abnormalities noted on inspection of the oropharynx.
Mild abnormalities were detected on laboratory results, including: white blood cell count 10.38 K/μL; hemoglobin 17.8 g/dL; hematocrit 53.4; platelet count 468 K/μL; blood urea nitrogen 15 mg/dL; creatinine 1.0 mg/dL; lipase in the normal range and a negative urine pregnancy test. The patient’s elevated hemoglobin and hematocrit within this clinical context was interpreted to be evidence of hemoconcentration and dehydration. Only yeast was detected on urinalysis.
Given the recurrent nature of her symptoms (four episodes within a year) and her abdominal exam findings, a computed tomography (CT) scan of the abdomen and pelvis with oral and intravenous contrast was obtained. The CT revealed dependant ascites in the abdominal and pelvic peritoneal cavities and discontinuous mural thickening in the proximal and middle small bowel suggestive of intestinal angioedema (Figures 1 and and 2).
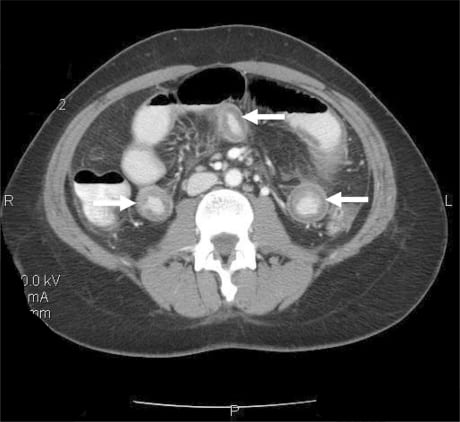
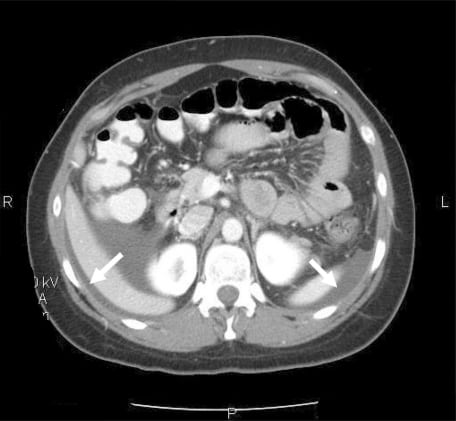
Upon review of the patient’s archived medical records, it was noted that she had previously been seen in the allergy clinic in 2004 with complaints of hand, foot and facial swelling. Biochemical testing performed at that time revealed a decreased C1 esterase inhibitor protein level of 5 mg/dL, and she was diagnosed with HAE and prescribed Danazol for prophylaxis against future attacks. However, the patient did not fill the prescription and was lost to follow-up. She had never previously experienced gastrointestinal symptoms related to her disorder prior to the past year and had not made the connection between her current symptoms and her diagnosis of HAE.
The patient’s allergist was called and notified of the patient’s presentation to the ED. He agreed that the totality of symptoms and clinical findings supported a diagnosis of an acute exacerbation of HAE, and an appointment was set within one week for further evaluation, treatment and definitive therapy. The patient reported that her symptoms had completely abated upon re-evaluation, and she was discharged home with a prescription for promethazine and instructions to consume clear liquids and advance her diet as tolerated for symptomatic control of her nausea.
DISCUSSION
HAE is a rare autosomal dominant disease first described by Sir William Osler in 1888.11 The disease is most commonly caused through a quantitative deficiency (type 1) or alternately due to a qualitative dysfunction (type 2) of the C1 esterase inhibitor protein in the plasma, which inhibits the action of vasodilators, such as kallikrein and bradykinin. Without inhibition from C1 esterase, bradykinin serves to increase vascular permeability resulting in angioedema that can occur at any mucosal or subcutaneous surface in the body.1
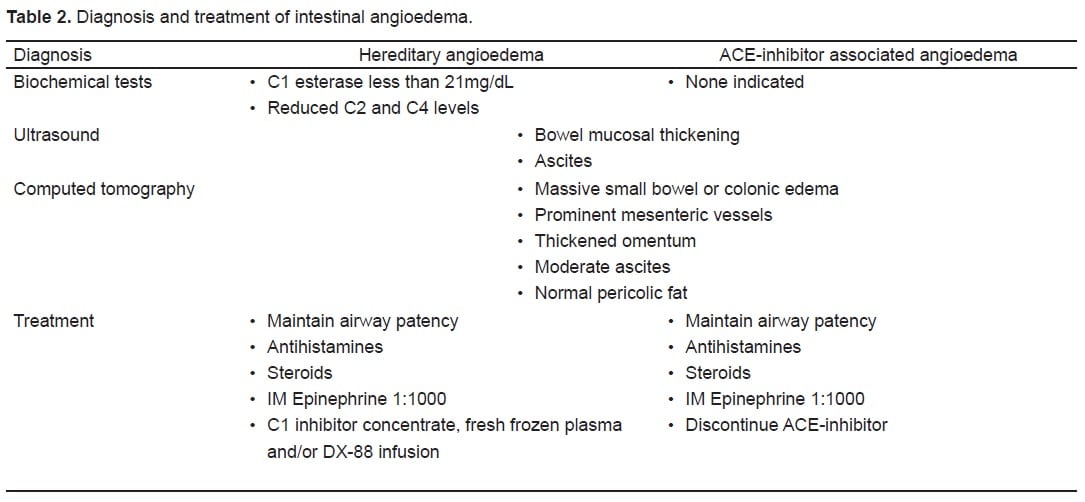
Although the patient in this case was suffering from a hereditary cause of her symptoms, the EP is more likely to encounter patients with angioedema due to ACE inhibitor use. However, angioedema from both etiologies presents similarly and share a common mechanism. ACE-inhibitors cause an excess of bradykinin by inhibiting its breakdown.12 Patients sensitive to increased levels of bradykinin will often only complain of a persistent, dry nagging cough, which resolves upon discontinuation of the offending medication, but in rare circumstances, bradykinin excess precipitates angioedema. The clinical findings and treatment of intestinal angioedema are summarized in Tables 1 and and 2.

Classically, HAE and ACE-inhibitor associated angioedema is located in the pharynx, extremities, or face. However, the bowel wall may be involved concomitantly in up to 75% of cases. Rarely, as we present in this case, angioedema of the bowel mucosa may be the only site of angioedema.2,13 The initial clinical presentation in patients with isolated gastrointestinal involvement may include colicky abdominal pain accompanied by nausea and vomiting that may easily be mistaken for appendicitis or biliary obstruction.2 About a day into an acute attack, the patient may experience watery diarrhea secondary to extravasation of fluid into the intraluminal space.2
Imaging with ultrasound or CT confirms the diagnosis of intestinal angioedema. Ultrasonography often reveals mucosal thickening and free peritoneal fluid in dependant areas of the abdomen, such as Morrison’s pouch.4 The sonographer may also detect a compressible but edematous bowel wall with increased intraluminal fluid and decreased motility.5 CT findings typically include massive edema of the small bowel or colon, prominent mesenteric vessels, thickened omentum, moderate ascites, and a normal appearance of the pericolic fat. Normal appearance of pericolic fat is useful for ruling out inflammatory changes seen in other diseases, such as appendicitis or diverticulitis.6,7Other findings may include increased contrast enhancement of the small bowel and mucosa with visualization of additional layers of the small bowel wall.8 Such findings are usually transient and will resolve following an acute episode of HAE or ACE-inhibitor associated angioedema.8,10 The appendix and gallbladder are usually well visualized, which may serve to effectively rule out some surgical etiologies if they are not involved.6 The differential diagnosis suggested by the findings on a characteristic CT for angioedema involving the bowel can be rather narrow and include ischemia and intramural bleeding secondary to coagulopathy or trauma.2
ACE-inhibitor induced angioedema is a clinical diagnosis, but use of diagnostic tests to determine the cause of angioedema in the ED is limited. If HAE is suspected, the diagnosis can be confirmed with a C1 esterase inhibitor level, although this may not practical or available in the ED setting. If performed, the normal limits of C1 esterase typically range from 21–39 mg/dL. A low C1 esterase inhibitor level is consistent with a diagnosis of HAE. In addition reduced levels of C2 and C4, which are substrates of the C1 esterase enzyme, further enhance specificity.1,2,3.
The management of this rare disorder in the ED remains largely undefined. Intestinal angioedema may be a particular challenge due to its rarity and the broad differential associated with its presentation. Unrecognized and untreated cases of angioedema exacerbations as a whole carry a mortality rate of 30–56%.5,13 Airway patency should be a priority in cases involving the oropharyngeal structures. When acute abdominal symptoms are present, surgical consultation may be prudent or required if there are doubts about the etiology of the clinical and radiologic findings, particularly when the patient is experiencing hemodynamic instability.10 In terms of immediate management, administration of C1 inhibitor concentrate and fresh frozen plasma have been reported to be effective in rapid reversal of some attacks.13–15 Cases related specifically to ACE-inhibitor associated angioedema are primarily treated through recognition and discontinuation of the offending agent. Antihistamines, steroids and intramuscular epinephrine 1:1000 may be used as adjunct therapies in cases where edema threatens airway patency. However, their efficacy has not been tested in clinical trials and is based on anecdotal reports. Recently, infusion of DX-88, a synthetic, potent inhibitor of kallikrein, has been successfully applied to accelerate resolution of attacks.9 Long term management for either etiology should include permanent discontinuation of ACE-inhibitors and close referral to an allergy and immunology specialist. For cases of HAE, androgen derivatives have been successful in terms of prophylaxis.13,15 Danazol is perhaps the most commonly prescribed agent, and tranexamic acid has been reported to be effective in terms of symptomatic treatment of swelling in up to 70% of patients.13,15
CONCLUSION
Angioedema secondary to HAE or ACE-inhibitor use is classically associated with facial and tongue swelling. However, intestinal angioedema commonly occurs in combination with facial involvement and rarely may occur in isolation. EPs must remain mindful of this entity, particularly in patients on ACE inhibitors or with a history of HAE presenting with acute abdominal pain, vomiting, or diarrhea. Early diagnosis may prevent morbidity from unnecessary exploratory surgery. Treatment of ACE-inhibitor-associated angioedema consists mainly of cessation of the offending agent; the management of HAE is much more poorly defined and should at minimum consist of supportive care and consultation with an allergy and immunology specialist.
Footnotes
Supervising Section Editor: Scott E. Rudkin, MD, MBA
Submission history: Submitted September 15, 2009; Revision Received January 8, 2010; Accepted February 12, 2010
Full text available through open access at http://escholarship.org/uc/uciem_westjem
Address for Correspondence: Edward J. LoCascio, MD, Department of Emergency Medicine, Louisiana State University Health Sciences Center-Shreveport, 1501 Kings Hwy, Shreveport, LA 71103
Email elocas@lsuhsc.edu
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding sources, and financial or management relationships that could be perceived as potential sources of bias. The authors disclosed none.
REFERENCES
1. Feller EJ, Spiro HM, Katz LA. Hereditary angioneurotic oedema: an unusual case of recurring abdominal pain. Gut. 1970;11:983–8. [PMC free article] [PubMed]
2. Ciaccia D, Brazer SR, Baker ME. Acquired C1 esterase inhibitor deficiency causing intestinal angioedema: CT appearance. AJR Am J Roetgenol. 1993;161:1215–6.
3. Hara T, Shiotani A, Matsunaka H, Yamanishi T, Oka H, Ishiguchi T, Saika A, Itoh H, Nishioka S. Hereditary angioedema with gastrointestinal involvement: endoscopic appearance. Endoscopy.1999;31:322–4. [PubMed]
4. Sofia S, Casali A, Bolondi L. Sonographic findings in abdominal hereditary angioedema. J Clin Ultrasound. 1999;27:537–40. [PubMed]
5. Burghardt W, Wernze H. Abdominal sonography in hereditary angioneurotic edema. A contribution to the early diagnosis of a disease of interdisciplinary significance. Ultraschall Med.1987;8:278–82. [PubMed]
6. Koruth JS, Eckardt AJ, Levey JM. Hereditary angioedema involving the colon: endoscopic appearance and review of GI manifestations. Gastrointest Endosc. 2005;61:907–11. [PubMed]
7. Wakisaka M, Shuto M, Abe H, Tajima M, Shiroshita H, Bandoh T, Arita T, Kobayashi M, Nakayama T, Okada F, Mori H, Uemura N. Computed tomography of the gastrointestinal manifestation of hereditary angioedema. Radiat Med. 2008;26:618–21. [PubMed]
8. De Backer AI, De Schepper AM, Vandevenne JE, Schoeters P, Michielsen P, Stevens WJ. CT of angioedema of the small bowel. AJR Am J Roetgenol. 2001;176:649–52.
9. Zingale LC, Zanichelli A, Deliliers DL, Rondonotti E, De Franchis R, Cicardi M. Successful resolution of bowel obstruction in a patient with hereditary angioedema. Eur J Gastroenterol Hepatol.2008;20:583–7. [PubMed]
10. Adhikari SP, Schneider JI. An unusual cause of abdominal pain and hypotension: angioedema of the bowel. J Emerg Med. 2009;36:23–5. [PubMed]
11. Osler W. Hereditary angioneurotic oedema. Am J Med Sci. 1888;95:362–7.
12. Ebo DG, Stevens WJ. Hereditary angioneurotic edema: review of the literature. Acta Clin Belg.2000;55:22–9. [PubMed]
13. Cicardi M, Bergamaschini L, Marasini B, Boccassini G, Tucci A, Agostoni A. Hereditary angioedema: an appraisal of 104 cases. Am J Med Sci. 1982;284:2–9. [PubMed]
14. Warrier MR, Copilevitz CA, Dykewicz MS, et al. Fresh frozen plasma in the treatment of resistant angiotensin-converting enzyme inhibitor angioedema. Ann Allergy Asthma Immunol.2004;92:573–5. [PubMed]
15. Goti F, Melcher GA, Späth P, Wüthrich B. Hereditary angioedema. A rare cause of acute abdominal pain with ascites. Dtsch Med Wochenschr. 1998;123:1166–71. [PubMed]