
{kind=link}
| Author | Affiliation |
|---|---|
| Ibrahim Abdullah, MD | St. Luke’s Hospital and Health Network, Bethlehem, PA |
| Kori Cossey, DO | St. Luke’s Hospital and Health Network, Bethlehem, PA |
| Rebecca K. Jeanmonod, MD | St. Luke’s Hospital and Health Network, Bethlehem, PA |
ABSTRACT
A 17-year-old male with symptoms of headache and diaphoresis presented to the emergency department. He had eight months of noted hypertension attributed to medications. On arrival his blood pressure was 229/117mmHg, and he was ill-appearing. His blood pressure was managed aggressively, and he was diagnosed with extra-adrenal pheochromocytoma by computed tomography. He eventually underwent resection of the mass. Children with severe, symptomatic hypertension should be evaluated for pheochromocytoma. Although rare, it is curable. Failure to diagnose carries a high risk of morbidity and mortality.
CASE PRESENTATION
A 17-year old African-American male presented to his school nurse with sudden onset of a sensation of all-over warmth, nausea, diaphoresis, mild headache, palpitations and shortness of breath. These symptoms occurred while sitting in class. On the nurse’s evaluation, the patient had a blood pressure of 260/140mmHg, so he was transferred to the emergency department (ED).
Upon arrival to the ED, the patient’s temperature was 97.6°F, his heart rate was 103 beats/min, respiratory rate 17 breaths/min, room air oxygen saturation 100% and blood pressure 229/117mmHg. His rhythm strip showed sinus tachycardia with what was believed to be likely sinus arrhythmia. He was diaphoretic and appeared dyspneic and ill. He had no thyromegaly, and pulmonary, abdominal, and neurological exams were all normal. His cardiac exam was remarkable only for tachycardia. He had no papilledema.
The patient had a past medical history of attention deficit hyperactive disorder for which he took 80mg of atomoxetine (norepinephrine uptake inhibitor) daily. He had been on this medication for several months. Previously, he had taken methylphenidate, but this had been stopped for unclear reasons. The patient had been noted to be hypertensive during a sports physical at school ten months previously, with a blood pressure of 180/100mmHg. He had undergone evaluation at that time with ultrasound of his kidneys and retroperitoneum, which showed no evidence of masses or renal artery stenosis. During the ensuing ten months, the patient had intermittent episodes of lightheadedness, headaches, diaphoresis and palpitations occurring one to two times per week and lasting between 15 and 60 minutes. He had been evaluated in the ED four months prior for lightheadedness, and at that time, the patient had a blood pressure of 159/99mmHg. He reported taking quetiapine and marijuana recreationally, and his elevated blood pressure was attributed to illicit substance use. His electrocardiogram (ECG) and chest radiograph had been normal, and his symptoms resolved spontaneously in the ED. On discharge, his blood pressure had been 141/91mmHg.
On his second ED visit, the patient’s differential diagnosis included pheochromocytoma, thyroid disease, renovascular hypertension and toxin-induced hypertension. Ancillary tests were done to narrow down this list. The patient’s ECG showed a sinus tachycardia with possible left ventricular hypertrophy (Figure 1). His chest radiograph was normal. His complete blood count, electrolytes, renal function and thyroid stimulating hormone were normal. His urine drug screen was negative. The patient was noted to have a mildly elevated calcium level (10.3mg/dL).
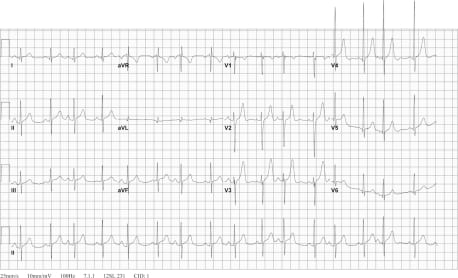
Based on the studies performed and presentation, the working diagnosis for the patient was then pheochromocytoma. Because the patient’s hypertension seemed the likely etiology of his symptoms, he was treated with 10mg of intravenous labetalol and 0.5 inches of 2% nitroglycerine paste. These were chosen because of their immediate availability in the ED. This resolved the patient’s symptoms. His blood pressure subsequently decreased to 144/86mmHg and his heart rate decreased to 71 beats/min. An echocardiogram and a computed tomography (CT) of the chest, abdomen and pelvis were ordered to look for possible etiologies of the hypertension, and he was admitted to the intensive care unit.
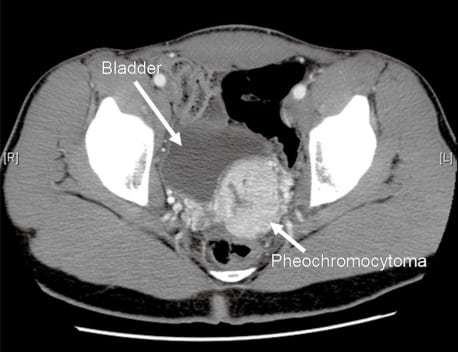
The patient’s echocardiogram showed evidence of concentric left ventricular hypertrophy, with no other abnormalities. His CT scan showed a 5.6 × 4.7 cm mass in the left pelvis along the posterior dome of the bladder, which was consistent with a pheochromocytoma, given its location and the clinical picture (Figure 2). Supporting inpatient lab studies revealed a plasma normetanephrine level of 4,062 pg/ml (normal is 0–145) and a urine norepinephrine of 3,304 ug/24 hours (normal is 0–140). Throughout the first two days of his hospital stay, the patient complained of frequent headaches. CT and magnetic resonance image (MRI) of the head were ordered to exclude metastasis to the brain or intracranial bleed, both of which were negative. He was started on phenoxybenzamine while an inpatient to control his blood pressure, to which he responded well.
The patient was discharged on phenoxybenzamine and remained using it for six weeks. He then underwent surgical resection of his pheochromocytoma. The mass showed evidence of vascular invasion and cell spindling, with three mitotic figures out of ten cells, which increases the malignant potential of his mass. The patient had a partial cystectomy and removal of his left vas deferens. His pathological margins were negative, and all resected lymph nodes showed no evidence of metastases. He was discharged after a brief stay in the hospital, during which he was hemodynamically stable. He was scheduled to follow up with endocrinology and hematology/oncology.
DISCUSSION
Although essential hypertension is increasing in incidence in children (likely secondary to obesity), all hypertension in the pediatric population should be considered secondary to organic disease rather than essential until proven otherwise.1 Hypertension in children is defined as a blood pressure greater than the 95th percentile for age, and is approximated by the formula 70 + (2 × age). Children presenting with higher blood pressures or at a younger age are more likely to have secondary causes of hypertension. Appropriate screening for and treatment of these diseases may reduce risk of long-term medical complications related to hypertension.1 Apart from pheochromocytoma, other reasons for secondary hypertension in children include primary renal disease (such as sponge kidney), renovascular disease (such as renal artery stenosis or thrombosis), congenital heart disease (such as coarctation of the aorta), pulmonary disease (such as bronchopulmonary dysplasia) and other forms of endocrine disease (i.e. hyperthyroidism), with renal causes being the most common.1 Most of these etiologies can be excluded using historical and physical findings, as well as routine blood tests, renal ultrasound and echocardiography. If these causes for hypertension are eliminated, pheochromocytoma must be ruled out in the pediatric patient.
Pheochromocytoma is a paraganglioma, or tumor, that arises from neural crest tissue. Paragangliomas may be parasympathetic or sympathetic in origin, but all pheochromocytomas are derived from chromaffin cells and are therefore sympathetic.2 Because of their embryological origin, they are found anywhere along the sympathetic chain, from the base of the brain to the urinary bladder. Pheochromocytoma is rare, accounting for 0.05–2% of all cases of hypertension, and there is no gender or race predilection.3 Seventy-five percent of cases are sporadic.4 In sporadic cases, the “rule of 10s” applies: 10% are bilateral, 10% are malignant, 10% are extra-adrenal, and of those, 10% are extra-abdominal. Familial cases (most commonly associated with the multiple endocrine neoplasia 2A and 2B syndromes, neurofibromatosis and Von-Hippel-Lindau syndrome) are bilateral half the time and malignant in 36% of patients.5 As many as 20% of newly diagnosed pheochromocytomas are in the pediatric population, in whom the disease is more commonly associated with these inherited syndromes.6–8 Extra-adrenal pheochromocytomas have a higher risk of malignancy than their adrenal counterparts (36%).5 While even “benign” pheochromocytoma is fatal if undiagnosed and untreated, it is potentially curable if diagnosed. Unfortunately, diagnosis is difficult and is typically delayed by about a year even in symptomatic children.9 Approximately 50% of cases are discovered at autopsy.10
The clinical manifestations seen in pheochromocytoma are due to the unregulated secretion of excess catecholamines, mainly norepinephrine.11,12 This activates alpha-adrenergic receptors in vascular smooth muscle, causing hypertension. Vascular constriction in the skin, gastrointestinal tract, brain, and kidney further potentiate systemic hypertension.11,12 Beta-adrenergic receptor activation also occurs, leading to increased cardiac output, with both chronotropic and inotropic effects. Beta activation increases renin secretion and worsens systemic hypertension via increased production of angiotensin II, antidiuretic hormone and aldosterone.11,12 Furthermore, insulin secretion is suppressed while glucagon secretion increases, leading to glycogenolysis, gluconeogenesis and lipolysis.13 The hypertension may be paroxysmal or sustained. Ultimately, malignant hypertension ensues leading to myocardial infarction, congestive heart failure, stroke, intracranial hemorrhage, papilledema or renal failure. If untreated, cardiovascular collapse and death follow.
The most common presenting symptoms in both adult and pediatric patients with pheochromocytoma are headache (70–90%), palpitations (50–70%) and diaphoresis (60–70%).7,9,14 Other common symptoms include anxiety, chest pain, dyspnea, fatigue, heat intolerance and focal neurologic deficits.7,14,15 The vast majority will have hypertension on exam (90–100%).14 Fever, pallor, hyperglycemia and vomiting are also commonly seen.9,14
Because the signs and symptoms of a pheochromocytoma can fit the clinical picture for many other diseases, the first and most important step in making the diagnosis is considering the disease. However, once pheochromocytoma enters the differential list, diagnosis can still be quite difficult as there is no one “best test.” Fractionated plasma metanephrines is considered the best initial screening exam, especially in high risk patients (i.e., those with positive family histories, classic histories or multiple endocrine tumors).16 This test can be skewed by certain medications, specifically tricyclic antidepressants and may be falsely elevated in the older patient population. A positive test should be confirmed with the highly specific 24-hour total urine metanephrine level.16,17 Despite the diagnostic use, the test is cumbersome and compliance is low, especially in children. Additionally, the clonidine suppression test, which measures plasma norepinephrine and metanephrines after a 0.3mg dose of clonidine, has been shown to differentiate between true and false elevations in plasma metanephrine levels.18 Unfortunately, none of these tests are practical in the ED.
If the provider is strongly considering a diagnosis of pheochromocytoma, admission for diagnostic studies is warranted. Asymptomatic, compliant adult patients with excellent follow-up could potentially be evaluated as outpatients, but symptomatic patients and children should be admitted. Once biochemical testing suggests the presence of a tumor, imaging studies should be ordered to locate it. Tumors greater than 3 cm can easily be identified on CT or MRI, yet both tests have their respective downfalls. CT often require contrast and has a higher radiation exposure, while MRIs are more expensive and have inferior spatial relationship when compared to CT.19 If no tumor is noted on preliminary imaging and clinical suspicion is still high, an I-MIBG study should be ordered. I-MIBG is a compound that resembles norepinephrine and is taken up by active adrenal tissue. Scans have a sensitivity of 77–90% and specificity of 95–100%.17,19 Finally, while small studies have shown positron emission tomography to detect 100% of tumors, cost and availability have prevented this from becoming the first line choice.19
Well before the final diagnosis of pheochromocytoma is made, urgent symptomatic treatment should be started with a goal of controlling severe hypertension and tachycardia. Definitive treatment is surgical removal of the tumor; however, medical management pre-and intra-operatively is extremely important. In the ED this should consist of initial treatment with an alpha blocker or vasodilator, such as nitroprusside or hydralazine, with addition afterwards of a beta blocking agent to control heart rate. Labetalol is also an appropriate first-line choice, as it has both alpha-and beta-blocking effects. Single drug therapy with a beta-blocker should be avoided because of the risk of severe, possibly fatal, hypertensive episodes caused by unopposed alpha-receptor stimulation.20 It is important to remember that, in spite of the hypertension, these patients require intravenous fluid resuscitation. Because of long-standing vasoconstriction, they can be profoundly dehydrated, so it is appropriate to give several liters of fluid with reassessment while treating the hypertension. Once the patient is stable, first-line medical therapy is an oral alpha-adrenoceptor blocker, the most effective of which is phenoxybenzamine. Phenoxybenzamine is an irreversible, long-acting alpha-adrenoceptor blocker that opposes extreme vasoconstriction caused by catecholamine surge.4 An oral beta-blocker can be added after adequate alpha-blockade is achieved. Lastly, metyrosine is another option for medical management, as it works through inhibition of catecholamine synthesis by blocking tyrosine hydroxylase. This medication has been shown to decrease clinical symptoms but can cause sedation, anxiety and diarrhea.20
All children with pheochromocytoma should be evaluated for inherited endocrine syndromes. They should all have long-term follow up to detect recurrence and metastases as early as possible. Most patients with appropriate surgical therapy have normalization of their blood pressure, but some will require long-term medical therapy, as well.
In conclusion, pheochromocytoma, while extremely rare, is a diagnosis that should be considered in the ED in pediatric patients with hypertension, particularly in those with malignant hypertension. Although it is not necessary (or sometimes even possible) for the emergency physician to make the diagnosis definitively, it is critical that the provider consider this entity and facilitate timely and appropriate evaluation. The emergency medicine provider must also begin an appropriate medical treatment regimen while avoiding potentially harmful therapies, such as beta-blocker monotherapy, in order to minimize morbidity and mortality from this disease.
Footnotes
Supervising Section Editor: Sean Henderson, MD
Submission history: Submitted June 17, 2010; Revision received September 13, 2010; Accepted October 11, 2010
Reprints available through open access at http://escholarship.org/uc/uciem_westjem.
Address for Correspondence: Rebecca Jeanmonod, MD, St. Luke’s Hospital and Health Network, 801 Ostrum St., Bethlehem, PA 18015
Email: rebeccajeanmonod@yahoo.com
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding sources, and financial or management relationships that could be perceived as potential sources of bias. The authors disclosed none.
REFERENCES
1. Brady TM, Feld LG. Pediatric approach to hypertension. Semin Nephrol. 2009;29(4):379–88.[PubMed]
2. Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the first international symposium. Nat Clin Pract Endocrinol Metab. 2007;3(2):92–102. [PubMed]
3. DeGraeff J, Horak BJV. The incidence of pheochromocytoma in the Netherlands. Acta Medica Scand. 1964;176:583–593.
4. Pacak K, Linehan WM, Eisenhofer G, et al. Recent advances in genetics, diagnosis, localization and treatment of pheochromocytoma. Ann Intern Med. 2001;134(4):315–29. [PubMed]
5. O’Riordain DS, Young WF, Jr, Grant CS, et al. Clinical spectrum and outcome of functional extraadrenal paraganglionoma. World J Surg. 1996;20(7):916–21. [PubMed]
6. Waguespack SG, Rich T, Grubbs E, et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2010;95(5):2023–37.[PubMed]
7. Hammond PJ, Murphy D, Carachi R, et al. Childhood phaeochromocytoma and paraganglioma: 100% incidence of genetic mutations and 100% survival. J Pediatr Surg. 2010;45(2):383–6.[PubMed]
8. Havekes B, Romijn JA, Eisenhofer G, et al. Update on pediatric pheochomocytoma. Pediatr Nephrol. 2009;24(5):943–50. [PubMed]
9. Bhansali A, Rajput R, Behra A, et al. Childhood sporadic pheochromocytoma: clinical profile and outcome in 19 patients. J Pediatr Endocrinol Metab. 2006;19(5):749–56. [PubMed]
10. Beard CM, Sheps SG, Kurland LT, et al. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950–1979. Mayo Clin Proc. 1983;58(12):802–4. [PubMed]
11. Williams RH, Foster DW, Kronenberg H, et al. Williams Textbook of Endocrinology. 9th ed. Philadelphia, PA: WB Saunders; 1998. pp. 665–728.
12. Pacak K, Lenders JWM, Eisenhofer G, et al. Pheochromocytoma: Diagnosis, Localization, and Treatment. Malden, MA: Blackwell Publishing; 2007. chapters 1–6.
13. Elenkova A, Matrozova J, Zacharieva S, et al. Adiponectin—a possible factor in the pathogenesis of carbohydrate metabolism disturbances in patients with pheochromocytoma. Cytokine.2010;50(3):306–10. [PubMed]
14. Manger WM, Gifford RW, et al. Clinical and experimental pheochromocytoma. 2nd ed. Oxford, United Kingdom: Blackwell Science; 1996. pp. 34–284.
15. Lai EW, Perera SM, Havekes B, et al. Gender-related differences in the clinical presentation of malignant and benign pheochromocytoma. Endocrine. 2008;34(1–3):96–100. [PubMed]
16. Sawka A, Jaeschke R, Singh RJ, et al. A comparison of biochemical tests for pheochromocytoma: measurement of plasma fractioned metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab. 2003;88(2):553–8. [PubMed]
17. Guller U, Turek J, Eubanks S, et al. Detecting pheochromocytoma: defining the most sensitive test. Ann Surg. 2006;243(1):102–7. [PMC free article] [PubMed]
18. Eisenhofer G, Goldstein D, Walther M, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true-from false-positive test results. J Clin Endocrinol Metab. 2003;88(6):2656–66.[PubMed]
19. Ilias I, Pacak K. Current approaches and recommended algorithm for diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab. 2004;89(2):479–91. [PubMed]
20. Adler J, Meyer-Rochow G, Chen H, et al. Pheochromocytoma: current approaches and future directions. The Oncologist. 2008;13(7):779–93. [PubMed]