
{kind=link}
| Author | Affiliation |
|---|---|
| Joe Lex, MD | Temple University School of Medicine, Department of Emergency Medicine, Philadelphia, Pennsylvania |
Xarelto® (rivaroxaban)
Eliquis® (apixaban)
Brilinta® (ticagrelor)
Abthrax® (raxibacumab)
Tudorza® (aclidinium bromide)
Sklice® (ivermectin)
Anascorp® (centruroides immune f(ab’)2 [equine])
Subsys® (fentanyl sublingual spray formulation for breakthrough cancer pain)
Dificid® (fidaxomicin)
Rectiv® (nitroglycerin ointment 0.4%)
Auvi-q®
Zio-patch®
ABSTRACT
To be honest, I thought this would be a lost cause. Even after skipping a New Drugs and Devices essay in 2012, I figured that I would have to search long and hard to find 10 new things that emergency practitioners needed to know about. Although there were no true blockbuster medications for emergency physicians, I nonetheless found 10 medicines that we probably should know, along with a new device that may change the way we work up patients with palpitations, and a clever new delivery system for subcutaneous epinephrine.
XARELTO® (RIVAROXABAN)
This one you’re going to hear about. Rivaroxaban is an oral anticoagulant that inhibits both free and bound Factor Xa. It is highly selective for this factor and has a rapid onset of action, reaching therapeutic levels in less than 4 hours. By inhibiting Factor Xa, both intrinsic and extrinsic pathways of the blood coagulation cascade are affected; thus, thrombin formation is blocked and clots are less likely to develop. It does not however inhibit thrombin (activated Factor II), and has no effects on platelets. Rivaroxaban has a flat dose response across an eightfold dose range (5–40 mg), so it theoretically allows predictable anticoagulation without dose adjustments and coagulation monitoring. Its half-life requires it to be taken twice daily to be effective.
Early in 2011, the United States (U.S.) Food and Drug Administration (FDA) approved rivaroxaban for prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE) in adults undergoing hip and knee replacement surgery; later in the year, the FDA approved it for stroke prophylaxis in patients with non-valvular atrial fibrillation (AF). Then on November 2, 2012, rivaroxaban was approved for the treatment of patients with DVT and PE and for long-term treatment to prevent recurrence. In other words, we now have an oral agent we can start in the emergency department (ED) to treat stable outpatients diagnosed with venous thromboembolic disease (VTE); as a bonus, it requires no bridging with heparin and no long-term monitoring.
While we will not be prescribing rivaroxaban for its first 2 indications, we will definitely be encouraged to use it for this most recent indication. Already, full-color 8-page glossy ads are showing up in our journals and monthly specialty-specific newspapers. Let’s admit it – the prospect of treating a stable patient with VTE as an outpatient simply by writing a prescription is difficult to ignore.
Does it work? Yes, it is at least as effective as the routine regimen of low molecular weight heparin (LMWH) and warfarin. The EINSTEIN-DVT study for treatment and secondary prevention of VTE was an unblinded, randomized, noninferiority study comparing oral rivaroxaban alone (15 mg twice daily for 3 weeks, followed by 20 mg once daily) with subcutaneous enoxaparin followed by a vitamin K antagonist (usually warfarin) for 3, 6, or 12 months in patients with acute, symptomatic DVT. As so often happens in real life, the INR was in the therapeutic range (2.0 to 3.0) for only 57.7% of the time. The number of recurrent clots was similar in both groups and the principal safety outcome of major bleedings was not different.
In the EINSTEIN–Pulmonary Embolism Study rivaroxaban was also noninferior to usual care (LMWH and warfarin) as far as recurrent VTE and bleeding in patients with symptomatic PE .
While warfarin is dirt cheap, rivaroxaban costs $8–9 a day…compared to $25 to $50 a day for generic enoxaparin. Somehow, somewhere, someone will determine that this is “cost effective.”
One big downside: As with dabigatran (Pradaxa®), there is no specific antidote for rivaroxaban in an exsanguinating patient. An antidote is, however, in development. Rivaroxaban’s half-life is only 5–13 hours, so withholding it may be enough. One study used Prothrombin Complex Concentrates (PCC) (50 IU/kg) in 12 healthy patients and showed reversal of the prolonged prothrombin time. However this may not correlate with hemostasis or patient-centered improved outcomes. Recall the excitement generated in studies using Recombinant Factor VIIa to limit the size of hemorrhage in cerebral bleeding, but which had no effect on patient-oriented outcomes, such as survival. Nonetheless, a trial of PCC is warranted in the exsanguinating patient anticoagulated with rivaroxaban. Because of its high protein binding, dialysis will not help. Protamine and vitamin K would not be expected to help.
Rivaroxaban is just the first drug of the xaban category to be approved for outpatient therapy of VTE. Many more will soon follow: apixaban (Eliquis®) is now also available and has been used in Europe since May 2012. Betrixaban, edoxaban, and otamixaban are all in various stages of human trials.
ELIQUIS® (APIXABAN)
Apixaban is another direct factor Xa inhibitor that has been available in Europe since May 2012. It barely made it under the wire for 2012, being approved by the FDA on 28 December for reducing the risk of stroke and systemic embolism in patients with AF that is not caused by a heart valve problem.
ARISTOTLE was a head-to-head study of apixaban 5 mg twice daily versus warfarin in patients with AF involving 18,201 patients in 1,034 clinical sites in 39 countries. The primary efficacy outcome was stroke or systemic embolism, analyzed by intention to treat. The primary safety outcome was major bleeding in the on-treatment population. Apixaban was not inferior to warfarin in the primary endpoint, and was superior in avoiding major bleeding (a key secondary endpoint). Time within the therapeutic range was mean 62%. See Table 1 for results.

ARISTOTLE
The downside of apixaban is the same as for rivaroxaban. No reversal agent is available for the exsanguinating patient. A trial scheduled to be evaluated in March 2013 (Efficacy and Safety Study of Apixaban for the Treatment of Deep Vein Thrombosis or Pulmonary Embolism) will give us information about whether apixaban can be used for outpatient treatment of VTE. Then, of course, we will need to see a head-to-head trial of rivaroxaban versus apixaban versus warfarin.
BRILINTA® (TICAGRELOR)
Ticagrelor reversibly inhibits the platelet 2Y12 adenosine diphosphate receptor. It is a cyclopentyltriazolopyrimidine, similar to the antiplatelet thienopyridines clopidogrel (Plavix) and prasugrel (Effient), both of which bind irreversibly to the P2Y12 receptor. All 3 drugs are used for secondary prevention of stent thrombosis, cardiovascular death, and heart attack in patients with acute coronary syndrome (ACS).
Ticagrelor was approved by the FDA based on the Platelet Inhibition and Patient Outcomes (PLATO) study, which looked at 18,624 patients with ACS from 862 centers in 43 countries who had symptom onset within.
Define CrCl, CABG and PLATO as footnote of table 2 the previous 24 hours. Unlike CURE and PCI-CURE, the two major studies evaluating clopidogrel, PLATO included patients with ST elevation myocardial infarctions (MIs). Patients without ST-segment elevation had to have 2 of the following for study inclusion: ST segment changes indicative of ischemia; a biomarker indicative of myocardial necrosis; or a “risk factor” (Table 2). Patients with ST-segment elevation had to have persistent ST-segment elevation of at least 0.1 mV in at least 2 contiguous leads or a new left bundle-branch block, plus planned primary percutaneous coronary intervention (PCI).

Risk factors for inclusion in platelet inhibition and patient outcomes.
Exclusion criteria included high risk of bradycardia, contraindication to clopidogrel, fibrinolytic administration within 24 hours of randomization, use of a strong CYP3A4 inhibitor or inducer, and oral anticoagulant that could not be discontinued.
In PLATO, patients were randomized to ticagrelor or clopidogrel. Patients who had not been taking aspirin received a loading dose of 325 mg, followed by 75 to 100 mg once daily, or 325 mg once daily for 6 months after stent placement.
While follow-up occurred at 1, 3, 6, 9, 12, and 13 months, the primary outcome measure was a composite endpoint at 12 months of cardiovascular death, MI, or stroke (similar to the CURE trial: PCI-CURE added recurrent angina into the composite). Results are shown in Table 3.
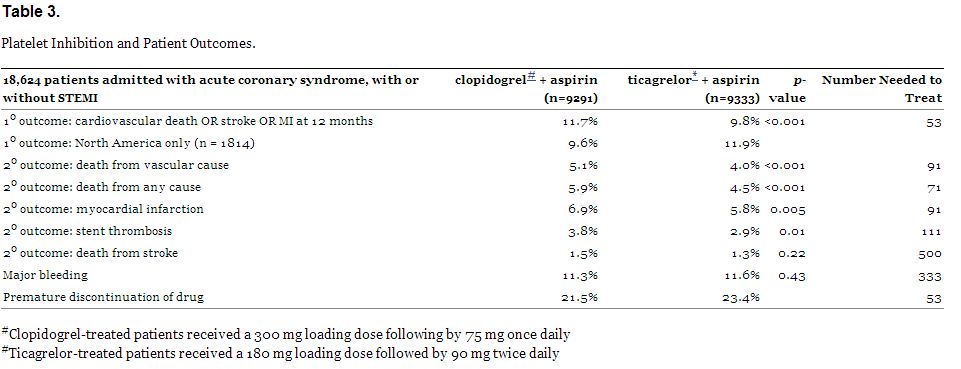
Platelet Inhibition and Patient Outcomes.
So for every 1,000 patients treated for up to 1 year with ticagrelor instead of clopidogrel, 11 fewer will suffer cardiovascular death, 11 fewer will have a heart attack, and 9 fewer will experience stent thromboses. The 1.9% absolute reduction (NNT=53) is being touted by AstraZeneca as a “16% reduction in death, stroke, or MI,” but that, of course, is a relative reduction.
In a strange finding, efficacy was lost for patients enrolled in North America (n=1814); in fact, clopidogrel proved superior to ticagrelor in reaching the primary endpoint (11.9% vs 9.6% – see Table 4). The reasons for this are unclear, but most North American patients were from the United States. U.S. patients were heavier, had more frequent co-morbidities and were more likely to undergo PCI or CABG.

Primary efficacy outcome, US vs non-US.
In addition, the median aspirin dose was higher in the U.S. – 325 mg versus 100 mg. Does this mean there is a drug interaction between aspirin and ticagrelor, or is it just that different aspirin doses reflect differences in patient populations. A post-hoc subgroup analysis suggested that aspirin dose, not geography, is to blame for the disparate results. Ticagrelor was approved with a requirement by AstraZeneca that they conduct educational outreach to physicians to alert them to the risk of using maintenance doses of aspirin over 100 mg daily, and there is a “black box” warning in the prescribing information about appropriate aspirin dosing.
In addition to bleeding, other adverse events included dyspnea (13.8% of patients) and, in those attached to a Holter monitor, ventricular pauses of more than 3 seconds (2%). Ticagrelor is a direct-acting oral antagonist of the adenosine diphosphate receptor, and some components of the molecular structure of ticagrelor are almost identical to those of adenosine; hence it is conceivable that metabolites of ticagrelor activate adenosine receptors. We know that adenosine induces dyspnea by bronchoconstriction, depresses the atrioventricular node, and causes deterioration in renal function by arteriolar constriction. These adverse events were generally self-limiting, but ticagrelor was discontinued because of adverse events more frequently than clopidogrel. Since less than 6% of the study patients had chronic renal disease, congestive heart failure, or obstructive pulmonary disease, we do not have good information on how these patients will fare taking ticagrelor.
Ticagrelor does not have to be metabolized to become active and thus has a faster onset than clopidogrel or prasugrel. In addition, it binds reversibly to the platelet, theoretically making it an attractive option in patients requiring surgery; nonetheless, the manufacturer recommends stopping the drug 5 days before major surgery, similar to clopidogrel.
In a striking coincidence, the average wholesale cost of the three most recent anticoagulant / antiplatelet drugs are similar (Table 5).

Some new anticoagulant /antiplatelet drugs.
ABTHRAX® (RAXIBACUMAB)
Raxibacumab is a newly approved drug for treatment of inhalational anthrax. It has the lumbering chemical formula C6320H9794N1702O1998S42. Like all monoclonal antibodies, the source and purpose are in the name. The last syllable (-mab) tells you that it is a monoclonal antibody. The antepenultimate syllable (-u-) tells you the source of the DNA is human (mouse DNA is designated by -o- and chimeric DNA by -xu-). And the preceding syllable (-bac-) tells you that it is an antibacterial agent.
Unlike antibiotics, which eradicate the Bacillus anthracis bacterium itself, raxibacumab targets the toxins produced by B. anthracis. These toxins are the cause of death in most human inhalational anthrax disease cases, including those in the 2001 attacks. As you may recall, as many as 68 victims were infected with inhalational anthrax delivered through the U.S. postal system, and 5 of them died despite aggressive antibiotic therapy.
More than 20,000 doses of raxibacumab have been in the Strategic National Stockpile since 2009. The stockpile includes other vaccines, antibiotics, and antitoxins that target anthrax and other agents of biological, nuclear and chemical warfare. Like raxibacumab, not all of these products are licensed.
Because naturally occurring inhalational anthrax infection in humans is rare (and studies deliberately exposing humans to the pathogen would be unethical), efficacy studies of raxibacumab were conducted in animals under the FDA’s Animal Rule. It is the first monoclonal antibody approved under this rule. Established in 2002, the Animal Rule allows developers to seek approval for the marketing of drugs or biologics based on efficacy data from animal studies, provided certain criteria are met. Safety data can be collected from humans, however, as was done with raxibacumab. Raxibacumab is administered in a single 40 mg/kg dose delivered intravenously over 2.25 hours. The instructions recommend premedication with diphenhydramine 1 hour before infusion.
Dividing anthrax bacilli produces protective antigen (PA), lethal factor (LF) and edema factor (EF). PA facilitates binding of LF and EF to anthrax toxin receptor (ATR) on mammalian cell surfaces, resulting in a protein-receptor complex that enables LF and EF to enter cells. These toxins inhibit normal immune system functioning, interfere with signal transduction pathways, and ultimately cause cell death. Antibiotics help control the bacterial infection, but they fail to clear toxins from the bloodstream. Vaccines can be effective over time, but they are slow acting initially and require booster doses to maintain immunity. By binding protective antigens, raxibacumab prevents LF and EF from entering cells, preventing progression of the disease.
Raxibacumab-treated animals had improved survival over control in 2 relevant animal models both in combination with antibiotics and alone. It is strictly for treating inhalational symptoms: Raxibacumab does not cross the blood-brain barrier and does not prevent or treat meningitis. Whether the drug will be dispensed solely by the U.S. government is unclear. I cannot find a charge for individual doses, but medicines supplied by the Strategic National Stockpile are free.
TUDORZA® (ACLIDINIUM BROMIDE)
We’ve known for many years that inhaled anti-cholinergic agents work as bronchodilators, and they are recommended as an option with beta-agonist therapy in most treatment algorithms for chronic obstructive pulmonary disease (COPD) and emphysema. Ipratropium bromide (Atrovent®), a short-acting quaternary ammonium compound, was standard therapy for many years, but its 4-time-daily dosing had fallen out of favor recently with the addition of once-daily tiotropium (Spiriva®) to the armamentarium. At first, aclidinium bromide (Tudorza®) seems like a step backward with its twice-daily dosing schedule.
There have been several published placebo-controlled trials of aclidinium, but very few head-to-head trials of aclidinium and another inhaled anticholinergic. The studies looked at data-oriented results – for instance, a 10% increase in FEV1 30 minutes after treatment; this improvement was noted in 49.5% treated with aclidinium, 51.8% treated with tiotropium, and 13.8% treated with placebo. Interestingly, a COPD symptom score was improved at night in users of the twice-daily aclidinium. This was thought due to the second, night-time dose. No attempt was made to compare twice-daily aclidinium to twice-daily tiotropium.
Many users of the tiotropium HandiHaler find it cumbersome, as it involves placing an intact capsule in a chamber, piercing it, and inhaling the contents. Aclidinium is a breath-actuated multi-dose dry powder inhaler, similar to the familiar Advair® Discus.
Lower cost is not an issue, as aclidinium costs $220 per monthly inhaler compared to $240 per month for tiotropium. Perhaps patients without dexterity to manage the somewhat tedious task of placing a capsule in the tiotropium dispenser would benefit with a switch to aclidinium, but there is no other clear reason for someone to switch.
SKLICE® (IVERMECTIN)
Pediculus capitis is a worldwide concern that affects persons of all socioeconomic backgrounds and ages, but it is most prevalent in children aged 3–13 years old. Since lice cannot fly or jump, transmission occurs through direct head-to-head contact, and possibly through the sharing of combs, hair brushes, or hats (although this is controversial). First-line treatments currently recommended by the American Academy of Pediatrics are the over-the-counter products, 1% permethrin or pyrethrins.
Permethrin (Nix®) has low toxicity, can be used in children as young as 2 months of age, and does not have cross-sensitivity with plant allergies (a theoretical risk with pyrethrins, which are derived from chrysanthemum flowers) However, resistance to permethrin is well documented and may limit its usefulness in certain areas of the country.
Pyrethrins (A-200, Licide, Pronto, RID, others) are neurotoxic to lice, but have little toxicity in humans. They can only be used in children 2 years of age and older and may cause an allergic reaction in patients with ragweed sensitivity. Neither permethrin nor pyrethrins are 100% ovicidal, since newly laid eggs do not have a nervous system for several days. Each costs about $20 for pediculocidal doses.
Product labeling of permethrin and pyrethrins recommends a second application at least 7 to 10 days after the initial application. Under average conditions, an egg or nit will hatch in approximately 8.5 days. Based on this time to hatch, a second treatment at 7 days will not be effective; some experts recommend the second treatment should we withheld until 9 to 10 days.
Lindane is no longer considered a first-line agent, as there are many reports of resistance, and it may have central nervous system side effects in humans. It should only be considered if head lice are unresponsive to other therapies, and then only in patients who weigh at least 50 kg. Its use has been banned in California.34
Malathion (Ovide®) is effective, but costs about $160. It is ovicidal, but the high alcohol content makes risk of accidental ingestion and flammability a concern. It is only approved for use in children 6 years of age and older, but resistance has not yet been proven in the U.S. (Malathion-resistance to lice is common in England.)
Benzyl alcohol lotion (Ulesfia®, which costs about $160, depending on hair length) is a suffocation-based therapy for head lice. It avoids pesticide or neurotoxin use, and resistance is not a problem, since it suffocates the lice. It can be used in children as young as 6 months of age, but kills only lice, and not the nits, so a second application is necessary 10 days after the initial application.
In the 12 years I have been doing this drug review, this will be the third unique product that I have reviewed for head lice. Topical ivermectin (Sklice®) is the newest product to be approved in the U.S. Ivermectin binds to glutamate chloride channels in nerve and muscle cells of lice, leading to an increased permeability to chloride ions resulting in paralysis and death. Based on this mechanism, it would appear that ivermectin is not ovicidal. But in vitro studies show that all lice hatched from eggs exposed to ivermectin died without the need for a second treatment. In addition, many of these lice were unable to suck blood, indicating that ivermectin somehow affected their ability to feed. One treatment costs approximately $260.
ANASCORP® (CENTRUROIDES IMMUNE F(AB’)2 [EQUINE])
Scorpions are predatory arthropods in the class Arachnida. They have 8 legs and are easily recognized by the pair of grasping claws and the narrow, segmented tail, often carried in a characteristic forward curve over the back, ending with a venomous stinger. They range in size from 9 mm to 21 cm and are widely distributed over all continents, except Antarctica. There are 1,752 described species of scorpions, with 13 families recognized. Scorpions are known to fluoresce when exposed to certain wavelengths of ultraviolet light, such as that produced by a black light, due to the presence of fluorescent chemicals in the cuticle.
Scorpion venom has a fearful reputation, and about 25 species are known to have venom capable of killing a human being.
Anascorp® is an antivenin indicated for treatment of scorpion envenomations, but only if symptoms develop, such as loss of muscle control, roving or abnormal eye movements, slurred speech, respiratory distress, difficulty with swallowing, excessive secretions or any symptoms that may increase risk for aspiration or compromised airway. It is available in the U.S. in some hospitals in Arizona and Nevada, where the dangerous bark scorpion is found.
Centruroides immune F(ab’)2 equine has a mechanism of action similar to that of other available antivenin products. It is composed of F(ab’)2 fragments specific to the toxic venom of Centruroides scorpions. These fragments target, bind, and neutralize the toxic venom. This promotes elimination and redistribution of the toxin from body tissues.
Centruroides immune F(ab’)2 equine is dosed by the whole number of vials until symptoms are resolved. The approved dosage is t3 vials as soon as possible after symptoms are observed, but an additional vial may be administered every 30 to 60 minutes if symptoms remain. Pharmacokinetic properties are shown in Table 6.

Pharmacokinetics of Centruroides Immune F(ab’)2
The 6 clinical trials of Centruroides immune F(ab’)2 equine show resolution of clinically important signs of envenomation when compared to placebo/supportive care. Only one controlled trial of Centruroides immune F(ab’)2 equine has been conducted consisting of 15 pediatric patients that presented for treatment within 5 hours of scorpion envenomation. This trial demonstrated a significant reduction of blood venom levels and full resolution of clinical symptoms by 4 hours post dose versus supportive care with midazolam.
As per the manufacturer, the shelf life of the product is 2 years. The average number of vials required to complete treatment is 3.59. Average treatment cost is $7,820.40 for 3 vials and $10,427.20 for 4 vials.
SUBSYS® (FENTANYL SUBLINGUAL SPRAY FORMULATION FOR BREAKTHROUGH CANCER PAIN)
Fentanyl is, of course, an opioid agonist with many indications for pain treatment. It is approximately 100 times more potent than morphine, with 100 micrograms of fentanyl approximately equivalent to 10 mg of morphine in analgesic activity. It is highly lipophilic and easily penetrates the blood-brain barrier. This sublingual spray formulation is indicated only for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and who are tolerant to opioid therapy for their underlying persistent cancer pain.
Fentanyl was derived from metabolites of meperidine (pethidine) in 1960 by Paul Adriaan Jan, Baron Janssen, founder of Janssen Pharmaceutica. The highly prolific baron also discovered haloperidol (1958), droperidol, and etomidate, as well as diphenoxylate, the primary ingredient in the anti-diarrheal medication, Lomotil.
Fentanyl has been used for widespread palliative use since the 1990s. First on the market was the transdermal Duragesic® patch, followed by first quick-acting fentanyl formations in a transmucosal formulation, the Actiq® lollipop and Fentora® buccal tablets. Even the U.S. military is looking at transmucosal fentanyl for battlefield treatment of pain. Sublingual spray is a logical extension of these other delivery systems. Derivatives of fentanyl have also been developed for specific purposes: there is a summary of these derivatives in Table 7.

Analogs of fentanyl.
Fentanyl is currently considered one of the safest opioids on the market, and the least physically harmful to the body, especially with long-term or life-term use. Its therapeutic index is 270:1. Fentanyl’s major side effects (>10% of patients) include diarrhea, nausea, constipation, dry mouth, somnolence, confusion, weakness, and sweating. Despite it being a more potent analgesic, fentanyl tends to induce less nausea and less histamine-mediated itching in relation to morphine.
Healthcare professionals who prescribe Subsys® on an outpatient basis must first enroll in the Transmucosal Immediate Release Fentanyl Risk Evaluation and Mitigation Strategy TIRF REMS ACCESS program (see https://www.tirfremsaccess.com/TirfUI/rems/home.action) and comply with the requirements of the REMS to ensure safe use of SUBSYS. As with all opioids, the safety of patients using such products is dependent on healthcare professionals prescribing them in strict conformity with their approved labeling with respect to patient selection, dosing, and proper conditions for use. While you will probably not prescribe Subsys®, you will almost certainly see patients who take it.
DIFICID® (FIDAXOMICIN)
Clostridium difficile is the most common cause of infectious diarrhea in hospitalized patients in North America and Europe, where both the incidence and severity of the disease have increased alarmingly since 2000. In most patients with this infection, there is a history of antibiotic or antineoplastic use within the prior 8 weeks. Its outcome can be anything from mild diarrhea to potentially-fatal pseudomembranous colitis. When identified, cessation of antibiotic is sufficient for cure in ∼25% of victims.
Oral metronidazole and oral vancomycin (whose name is derived from the term “vanquish”) have similar efficacy for mild to moderate C. difficile infection. Metronidazole is preferred due to concerns about cost and the potential for vancomycin resistance. For severe infections, response rates to oral vancomycin are significantly better than with oral metronidazole (response rates 97% versus 76% and 85% versus 65% for vancomycin and metronidazole, respectively in 2 studies).
Fidaxomicin shows similar efficacy to vancomycin and may be a therapeutic option in mild to moderate cases of C. difficile diarrhea, but a 10-day course costs approximately $2,800, significantly higher than a 10-day course of oral vancomycin (∼$1300). If you reconstitute injectable vancomycin with sterile water and dilute it to a concentration of 50 mg/mL, then direct that it be used orally, possibly with flavoring syrup, the cost of a course of therapy is $60 or less.
The cheapest and apparently most effective treatment for infection with C. difficile appears to be fecal transplant, shown in several small series to completely correct the condition. This esthetically-disturbing treatment, described as early as 1958, is being used more and more frequently. It is also called fecal microbiota transplantation, or FMT. The procedure usually involves an infusion of bacterial fecal flora harvested from a healthy donor. The stool can be given by enema or colonoscopy, or through a nasogastric or nasoduodenal tube. Most patients recover clinically and have C. difficile eradicated after just 1 treatment. Donors should be tested for a wide array of bacterial and parasitic infections, including occult C. difficile.
Perhaps we should be encouraged to keep a healthy specimen of our own stool in refrigeration, as Autologous Restoration of Gastrointestinal Flora (ARGF) has also been recommended. Should you develop C. difficile, your stool flora are extracted with saline and filtered, then freeze-dried and placed in enteric-coated capsules, which you can take orally to restore your original colonic flora.
RECTIV® (NITROGLYCERIN OINTMENT 0.4%)
Most anal fissures are caused by stretching of the anal mucosa beyond its capability; in adults, this includes constipation, the passing of large, hard stools, prolonged diarrhea, or anal sex. Sometimes they cause bright red blood on the toilet paper, occasionally in the toilet. When acute they cause pain after defecation; chronic fissures cause pain less frequently. Anal fissures typically extend from the anal opening and are usually posteriorly in the midline, probably because of the relatively unsupported nature and poor perfusion of the anal wall in that location. Fissures can be superficial or extend down to the underlying sphincter muscle. The incidence of anal fissures is around 1 in 350 adults, equally common in men and women, and most frequent in young adults aged 15 to 40.
First-line management of anal fissure generally includes increasing fluid and fiber intake, stool softeners, topical analgesics (e.g., 1% lidocaine) or anti-inflammatories (e.g., 1% hydrocortisone), and sitz baths.
Nitroglycerin is a vasodilator and causes smooth muscle relaxation. It was first used by William Murrell to treat anginal attacks in 1878. As a topical agent, it is applied as a 0.2% to 0.4% ointment. When applied topically to the anus, it increases local blood flow, relaxes anal sphincter tone, and reduces anal pressure. The literature is mixed, but according to pooled data, topical nitroglycerin appears to be associated with healing in at least 50% of treated chronic fissures and is associated with a significant decrease in pain.
The recommended dosage is 1 inch of ointment (375 mg of ointment equivalent to 1.5 mg of nitroglycerin) applied intra-anally every 12 hours for up to 3 weeks. As with all nitrates, Rectiv® is contraindicated within a few days of using PDE5 inhibitors such as sildenafil, tadalafil, and vardenafil due to potentiating hypotensive effects. After treatment with topical nitroglycerin, recurrence of anal fissures occurs in about one-third of the patients over the following 18 months.
Topical calcium channel blockers such as diltiazem and nifedipine inhibit calcium ion entry through voltage-sensitive areas of vascular smooth muscle and also cause muscle relaxation and vascular dilatation. By relaxing the internal anal sphincter, calcium channel blockers also lower the resting anal pressure. In fact there is evidence that topical calcium channel blockers may be as effective as topical nitroglycerin, but with fewer side effects, such as headache. Diltiazem and nifedipine are not available in topical ointment or gel form and must be compounded by a pharmacist. The typical strengths used for anal fissure are diltiazem 2% and nifedipine 0.2% to 0.5%. The usual dosing is a pea-sized amount applied rectally 2 to 4 times daily.
Considering the ubiquity of nitroglycerin, the cost for a 30-gram tube is staggeringly expensive: US$386.
AUVI-Q®
This is a crossbreed between a drug, which is very old, and a device which is very new. First of all, do you call the drug adrenaline (or adrenalin) or epinephrine (or epinephrin)? It was in 1893 that George Oliver, a Harrogate physician, and Edward Schäfer, professor of physiology at University College London, demonstrated that the adrenal (or suprarenal) glands contained a substance with dramatic pharmacological effects. American physician John Abel named the crude adrenal extracts he prepared in 1897, calling them epinephrin, thinking that epinephris was the best name for the suprarenal capsule.
But none of Abel’s epinephrin extracts behaved physiologically like adrenaline does. Jokichi Takamine visited Abel in 1901; afterward, he prepared a pure extract of the active principle from the adrenal gland and patented it. When Parke, Davis & Co marketed his extract, they used the proprietary name Adrenalin. Thus, epinephrine became the generic name in America.
For patients with a history of anaphylaxis and severe allergy, auto-injectors containing a dose of epinephrine between 300 and 500 μg at a 1:1000 concentration (commercial names EpiPen, Twinject, Adrenaclick, Anapen, Jext, and Allerject) have been available for decades. They are somewhat cumbersome to carry because of their size and shape. In times of great stress, such as after having a potential allergic exposure, the patient often forgets the instructions given at the time of distribution.
Auvi-Q® is an epinephrine auto-injector that is amere 3.5 inches tall by 2 inches wide and as thick as a cell phone. This makes it easy to carry in the pants back pocket or shirt pocket. As an advantage, it “talks” to you when you remove the cover, walking you through the process of injection and counting down the number of seconds to leave the device in place. The cost is about the same as other commercially-available devices. Thus, I do not know that it offers any advantage over other auto-injectors, which generally retail for $255 for 2 devices. A common complaint about the older devices is that they expire so quickly without being used, and users may be tempted to squeeze another year or two out of them. A study done in 2000 shows that they actually do lose efficacy over time.
ZIO-PATCH®
In 1946, a physicist / chemist named Norman Jefferis “Jeff” Holter returned to his home of Helena, Montana, to establish a research lab after serving in the U.S. Navy Bureau of Ships and researching the behavior of waves. One of his early inventions was a device that still bears his name: the Holter monitor. In those pre-transistor days, the device was the size of a very large backpack and weighed about 85 pounds. The size of recorder varies among manufacturers, but the average dimensions of today’s Holter monitors are about 110×70×30 mm. Most of them operate with two AA batteries and record a continuous period of 24 to 48 hours. The first report of Holter monitoring in humans appeared in 1954 by MacInnis. If you go to PUBMED now and search “Holter monitor,” you will find more than 11,000 references.
The Zio® Patch is a single-use, noninvasive, waterproof, long-term continuous monitoring patch worn on the chest that provides continuous monitoring for up to 14 days. Theoretically by providing a longer time period of continuous recording, the Zio® Patch improves the likelihood of capturing arrhythmias and provides for an equal or higher diagnostic yield versus other devices on the market. Thus far there is very little literature on this device, but initial investigators gave it glowing reviews. While the authors acknowledged receiving a restricted research grant from the manufacturer iRhythm, they stated there were no conflicts of interest.
In their study they compared the Zio® Patch to a 24-hour Holter monitor in 74 consecutive patients with paroxysmal atrial fibrillation referred for Holter monitoring for detection of arrhythmias. The Zio® Patch allowed a mean monitoring period of 10.8 ± 2.8 days (range 4–14 days). Over a 24-hour period, there was excellent agreement between the Zio® Patch and Holter for identifying atrial fibrillation events and estimating atrial fibrillation burden. Atrial fibrillation events were identified in 18 additional individuals in the Zio® Patch group, prompting therapy change. Other clinically relevant cardiac events recorded on the Zio® Patch after the first 24 hours of monitoring included symptomatic ventricular pauses, which prompted referrals for pacemaker placement or changes in medications.
The patient can remove the patch after the observation time and mail it back to the physician. Cost is about $150 per patch, considerably less than the cost of Holter monitoring. I predict that when these become freely available, emergency physicians will jump at the opportunity to use them. Think about the number of patients we see complaining of palpitations or similar symptoms and how difficult it is to arrange a Holter monitor from the ED. With the Zio® Patch, we take a Band-Aid®-sized device out of its package and slap it on the patient’s chest and give them instructions about what to watch for and who to follow up with.
On the other hand, as with any innovative new device I worry about the “technological imperative,” also known as “the inevitability thesis.” Simply stated, “whatever can be done will be done,” or, to put it more bluntly, “once you are handed a hammer, everything starts to look like a nail.” Will we save lives with this device, or will we pick up “incidentalomas” that condemn patients to a lifetime of medication or, worse yet, internal defibrillators and pacemakers because now we can identify trivial problems that may never cause a symptom. Will we make our patients VOMIT (Victims Of Medical Investigational Technology)? We’ll have to see.
Footnotes
Address for Correspondence: Joe Lex, MD, Temple University. Email: joe@joelex.net 11 / 2013; 14:619 – 628
Submission history: Revision received February 20, 2013; Accepted February 22, 2013
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding sources and financial or management relationships that could be perceived as potential sources of bias. “This article was originally published on 1 March 2013 by www.EMedHome.com, where it was offered for 2 hours of CME.” The author disclosed no other potential sources of bias.