
{kind=link}
| Author | Affiliation |
|---|---|
| Eric F. Silman, MD | University of California, San Francisco |
| Bharath Chakravarthy, MD | Loma Linda University |
| Federico Vaca, MD, MPH | University of California, Irvine School of Medicine |
| Mark I. Langdorf, MD, MHPE | University of California, Irvine School of Medicine |
ABSTRACT
Autosomal dominant polycystic kidney disease may present to the emergency department (ED) with vomiting, abdominal pain or hernias, renal insufficiency or failure, or bleeding from cerebral aneurysms. A 37-year-old man presented to the ED with signs and symptoms of incarcerated inguinal hernia. Laboratory studies showed renal failure with anion gap acidosis, and bedside ultrasound showed multicystic kidneys. Computed tomography confirmed the diagnosis. Emergency physicians should be aware of this common connective tissue defect and its serious associated conditions.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is relatively common, estimated to occur in one in 1000 live births.1 A defect in epithelial cell function, its multiorgan structural manifestations cause complications including hepatic cysts, cerebral arterial aneurysms, and abdominal wall hernias, any of which are life-threatening and require prompt diagnosis and treatment in the emergency department (ED).
CASE
A 37-year-old Hispanic male presented with progressive pain in his abdomen and inguinal area for four days. The pain began suddenly and was located in the right upper quadrant, right inguinal and scrotal area. The patient had anorexia, nausea and vomiting the prior day with chills, fevers to 105°F and sweats. He had normal bowel movements without hematochezia or melena. He also reported a three-year history of inguinal hernia that was previously asymptomatic. Review of systems was significant for three episodes of gross hematuria in the previous three days without urgency, frequency, or changes in urinary output. All other review of systems was negative. The patient denied any medication use or allergies. There was no significant prior illness, surgery, or family medical history. He had not seen a doctor since childhood.
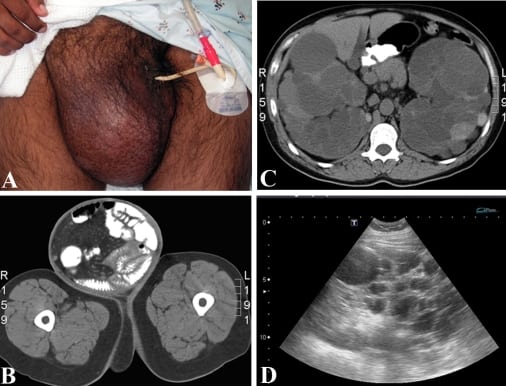
The patient’s initial vital signs were a temperature of 36.8°C, a heart rate of 94 beats per minute, a blood pressure of 137/86 mmHg, respirations at 18 breaths per minute and oxygen saturation of 99% on room air. Relevant physical findings included a soft and diffusely tender abdomen, with rebound tenderness and pain on gurney movement suggesting peritoneal irritation. There was an exquisitely tender 25 × 30 cm right inguinal hernia extending into the scrotum with bowel sounds and visible peristalsis, without overlying skin changes or crepitus (Figure 1A). There were no other masses appreciated on physical exam and the remainder of the physical examination, including rectal and genitourinary, was unremarkable.
Pertinent initial laboratory results included: a leukocytosis of 18,300 cells/mm3 with 84% neutrophils and no bands, a mild anemia with hemoglobin of 11.1 g/dl and a normal platelet count. Chemistry results yielded potassium of 3.3 mEq/dl, bicarbonate of 8 mEq/dl, anion gap of 20, blood urea nitrogen (BUN) of 178 mg/dl and creatinine of 19.9 mg/dl. Lipase was greater than 400 U/l with normal hepatic function panel. Urinalysis showed positive leukocyte esterase and negative nitrite, 20 mg% protein, large hemoglobin, 300 red blood cells and 23 white blood cells per high-power field. A surgical consultation was obtained to evaluate for strangulated inguinal hernia. Initial therapy included intravenous piperacillin/tazobactam in the ED.
Arterial blood gas revealed pH 7.10, pCO2 16 mmHg and bicarbonate 4.9 mEq/L with a base deficit of 23. Serum lactate level was normal, suggesting the anion-gap acidosis was more likely due to uremia than bowel infarction. A Foley catheter returned 50 ml amber-colored urine.
Abdominal CT showed intraluminal contrast in the hernia sac (Figure 1B), diffuse small bowel edema with a transition point at the entrance to the hernia sac, multiple liver cysts and bilaterally enlarged kidneys with innumerable cysts filling most of the abdominal cavity (Figure 1C), as well as a 7 mm stone in the left renal pelvis. The pancreas was incompletely seen due to lack of intravenous contrast, which was withheld due to renal failure. A bedside ultrasound performed in the ED showed multiple cystic structures in both upper quadrants and flanks (Figure 1D).
A dialysis catheter was placed in the ED and the patient was emergently dialyzed. He was admitted to intensive care for acute renal failure, incarcerated inguinal hernia with a partial small bowel obstruction, dehydration and an acute exacerbation of polycystic kidney disease. The patient was awake, alert, and in only moderate distress throughout his ED course, suggesting an element of tolerance to chronically elevated BUN and creatinine levels. Regarding family history, he reported that his father had a renal transplant for unknown reasons.
The following day the patient’s electrolytes and pH nearly normalized. His BUN/creatinine, white blood cell count and lipase trended down in the following days. Blood and urine cultures were negative. Urology was consulted and elected to delay nephrectomy to determine if one kidney could be salvaged. General surgery planned a hernia repair after the nephrectomy. The partial small bowel obstruction resolved with bowel rest. A screening MRI of the brain the following day revealed no bleeding or cerebral aneurysms.
DISCUSSION
The diagnosis of ADPKD may become evident to the emergency physician (EP) in four ways. First, routine testing for complaints of vomiting or abdominal pain may reveal renal failure. Second, abdominal wall or inguinal hernias associated with renal insufficiency or hematuria may suggest the diagnosis. Third, with increasing use of ED ultrasound, multicystic kidneys may be seen during a search for the cause of abdominal pain. Finally, non-contrast CT, while investigating abdominal or flank pain, or clinical bowel obstruction, may offer the unifying diagnosis. In our patient, surgery for incarcerated hernia with bowel obstruction was delayed by the condition which caused the hernia, namely renal failure from ADPKD.
ADPKD affects approximately one in 1000 people.2 Cysts arise from the nephrons and collecting tubules.2 The most common presentation is a palpable mass, hypertension (after their third decade of life), abdominal pain, and hematuria.1,2 Hypertension often predates renal failure. Up to 73% of patients will have hepatic cysts, 9% have pancreatic cysts, and 5% have splenic cysts.2–6 Family history often includes renal disease and/or transplantation, as in our patient, and abdominal CT scan shows bilaterally-enlarged kidneys with multiple cysts. It is the fourth leading cause of end-stage renal disease in adults,7 and about 10–12% of patients receiving maintenance hemodialysis have ADPKD.2–6
The genetic basis for ADPKD has been traced to mutations in two genes. Type I is caused by a defect in the PKD1 gene (85–90% of cases), while Type II is related to a defect in the PKD2 gene.8
Diagnostic criteria center on the presence and quantity of cysts, age, and familial risk. Patients typically present with enlarged kidneys with multiple cysts bilaterally and a positive family history consistent with autosomal dominant inheritance. For age 15–29, at least two cysts (uni- or bilateral) are sufficient for diagnosis. At age 30–59, as the prevalence of simple cysts increases, at least two cysts in each kidney are required for diagnosis, and beyond age 60, four cysts on each side. 9
The common endpoint in half of ADPKD patients is renal insufficiency.6 Concomitant cardiovascular pathology includes aortic root dilatation, aortic regurgitation, bicuspid aortic valves, coarctation of the aorta, mitral regurgitation, and abdominal aortic aneurysm.10, 11 Aneurysms of cerebral arteries have been found in up to 50% of patients.10–12 Abdominal wall hernias are up to five times more common in ADPKD patients with prevalence up to 45%.14 The prevalence of abdominal wall hernia is even higher in those on chronic ambulatory peritoneal dialysis.14 The most common type of hernias are inguinal followed by incisional and paraumbilical.13,14 The increased prevalence is thought to be due to a combination of increased intraabdominal pressure from enlarged kidneys and weak abdominal musculature due to the connective tissue pathology.12 We could find no data on the prevalence of incarceration or strangulation compared to the general population.
In addition to renal failure, this particular patient had laboratory and imaging evidence for pyelonephritis and nephrolithiasis, two other common complications of ADPKD.13Despite a significantly elevated serum lipase, pancreatitis was not diagnosed. The level was thought to be due to a combination of decreased renal excretion as well as intraperitoneal irritation. Pancreatic cysts can be present with ADPKD, but our patient had none.15
ADPKD has an array of associated symptoms and complications. Many of these are potentially life-threatening and may bring a patient to the ED. It is important for the EP to be familiar with these and not to be distracted from the true etiology.
Footnotes
Supervising Section Editor: James P. Killeen, MD
Submission history: Submitted November 08, 2008; Revision Received August 04, 2008; Accepted August 28, 2008.
Full text available through open access at http://escholarship.org/uc/uciem_westjem
Address for Correspondence: Mark I. Langdorf, MD, MHPE. Department of Emergency Medicine, University of California, Irvine School of Medicine, 101 The City Dr., Rte 128-01, Orange, CA 92868
Email: milangdo@uci.edu
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding sources, and financial or management relationships that could be perceived as potential sources of bias. The authors disclosed none.
REFERENCES
1. Gabow PA. Medical progress: Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. [PubMed]
2. Khan A, Chandramohan M, MacDonald S. Autosomal Dominant Polycystic Kidney Disease. eMedicine. 2007. [Accessed July 04, 2008]. Available at:http://www.emedicine.com/Radio/topic68.htm.
3. Higashihara E, Aso Y, Shimazaki J. Clinical aspects of polycystic kidney disease. J Urol.1992;147:329–332. [PubMed]
4. Itai Y, Ebihara R, Eguchi N. Hepatobiliary cysts in patients with autosomal dominant polycystic kidney disease: prevalence and CT findings. Am J Roentgenol. 1995;164:339–342. [PubMed]
5. Milutinovic J, Agodoa LC, Cutler RE. Autosomal dominant polycystic kidney disease. Early diagnosis and consideration of pathogenesis. Am J Clin Pathol. 1980;73:740–747.[PubMed]
6. Parfrey PS, Bear JC, Morgan J. The diagnosis and prognosis of autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323:1085–1090. [PubMed]
7. Langdorf MI, Rudkin SE, Dellota K, Fox JC, Munden S. Decision rule and utility of routine urine toxicology screening of trauma patients. Eur J Emerg Med. 2002;9:115–121. [PubMed]
8. Grantham JJ, Torres VE, Chapman AB. CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med. 2006;18:2122–2130. [PubMed]
9. Wilson PD. Polycystic Kidney Disease. N Engl J Med. 2004;350:151–164. [PubMed]
10. Pei Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2006;1:1108–1114. [PubMed]
11. Nakajima F, Shibahara N, Arai M. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol.2000;164:311–313. [PubMed]
12. Schievink WI, Torres VE, Piepgras DG. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1992;3:88–95. [PubMed]
13. Fick GM, Gabow PA. Hereditary and acquired cystic disease of the kidney. Kidney Int.1994;46:951–964. [PubMed]
14. Gabow PA. Autosomal dominant polycystic kidney disease — More than a renal disease. Am J Kidney Dis. 1990;16:403–413. [PubMed]
15. Morris-Stiff G, Coles G, Moore R. Abdominal wall hernia in autosomal dominant polycystic kidney disease. Br J Surg. 1997;84:615–617. [PubMed]
16. Royse VL, Jensen DM, Corwin HL. Pancreatic enzymes in chronic renal failure. Arch Intern Med. 1987;147:537–539. [PubMed]