
{kind=link}
| Author | Affiliation |
|---|---|
| Bradley C Presley, MD | Medical University of South Carolina, Division of Emergency Medicine, Charleston, South Carolina |
| Jeffrey S Bush, MD | Medical University of South Carolina, Division of Emergency Medicine, Charleston, South Carolina |
| Simon C Watson, MD | Medical University of South Carolina, Division of Emergency Medicine, Charleston, South Carolina |
ABSTRACT
This report reviews a case of dermatomyositis presenting with weakness and extensive calcification in an adult. While dermatomyositis is not uncommon in adults, it is uncommon for calcifications to be present. Children develop calcifications more frequently than adults. When present in adults, small calcifications on areas of frequent trauma such as elbows and fingers are more common. However, this patient presented with large calcified deposits in his abdomen and extremities. His treatment and course are described.
CASE REPORT
A 26-year-old Hispanic male presented to the emergency department with progressive weakness during the previous year. This weakness initially started in his lower extremities, when he noticed difficulty when ascending stairs; however, it progressed to involve his upper extremities in the following months. He also had complaints of joint pain, generalized fatigue, “bumps,” and “hard places” over his abdomen and thighs, as well as a rash that was more pronounced on his face, arms, and legs. Of note, he had been admitted to another hospital several months before and treated with aggressive hydration for rhabdomyolysis. He improved slightly after that admission, but the weakness subsequently progressed to the point that he was unable to ambulate on his own power. He was given a prescription for an unknown medication on discharge from his previous hospitalization but it was never filled. His past surgical history was significant for an appendectomy about 1 year prior; his family history was unremarkable.
On physical examination, the patient had normal vital signs. He was noted to have a diffuse hyperpigmented rash over his face, more prominent on his cheeks and other sun-exposed portions of his body. He was also noted to have taut skin and a firm, nonmobile, tender mass in his right lateral abdominal and flank area that began at the costal margin and continued into his right pelvis. This firm area extended from the posterior axillary line to almost the midline of his abdomen. He had other similar but smaller masses in his left upper and lower quadrants as well as on both thighs. On examination, the patient was unable to raise his arms above his head without assistance. His neurologic examination showed intact sensation and reflexes throughout with marked weakness in his extremities and trunk. He demonstrated 2–3/5 strength in both legs proximally with 4/5 strength in his upper extremities. He had difficulty sitting up without assistance.
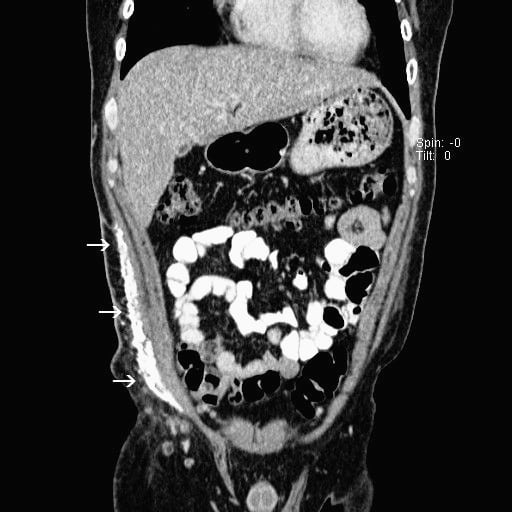
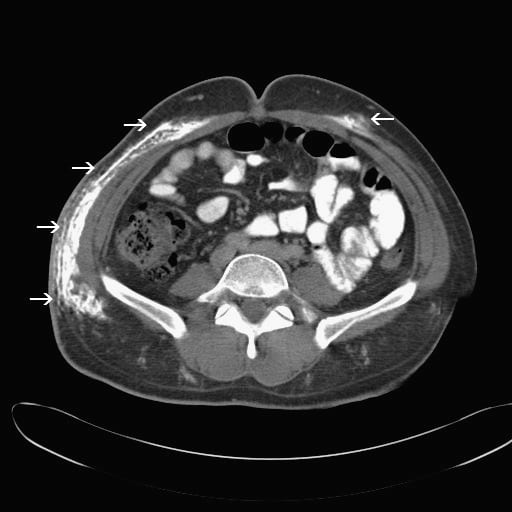
His laboratory tests in the emergency department included a basic metabolic panel that was within normal limits, with normal calcium levels. The creatine kinase (CK) level, however, was elevated to 3,501 IU/L, while serum aldolase was 29.7 U/L (reference, 1.5–8.1 U/L) and lactate dehydrogenase (LDH) was 728 IU/L (reference, 100–240 IU/L). He had a mild transaminitis with an aspartate aminotransferase level of 207 IU/L and an alanine aminotransferase level of 148 IU/L. A computed tomography of the abdomen and pelvis was completed. The official read commented on calcifications in the right rectus abdominis muscle, external oblique, and subcutaneous fat, as well as calcifications in the left external oblique muscle and left rectus abdominis. The patient was admitted for further management.
In the hospital, he was aggressively hydrated and given high-dose steroids as well as methotrexate (Trexall; Teva Pharmaceuticals USA, North Wales, Pennsylvania). Rheumatology service was consulted, and a muscle biopsy confirmed the diagnosis of dermatomyositis with calcinosis cutis. The patient was then given azathioprine (Imuran; Prometheus Laboratories Inc, San Diego, California) and he showed continual improvement throughout his hospital course. His CK levels improved with hydration, and he received intensive physical therapy throughout his hospitalization. At hospital discharge, the patient had 4/5 strength globally and was able to sit in a chair unassisted. He continues to work toward ambulation through outpatient physical therapy.
DISCUSSION
Dermatomyositis is a disease that can present in individuals of all ages, with peak incidence in adults during the fifth and sixth decades of life. It has an incidence of 5.5 per million people. The exact mechanism for dermatomyositis is not known, but it is postulated to have an autoimmune component. Some cases are felt to be paraneoplastic. In most cases, the rash and proximal weakness appear simultaneously. However, 30% of patients experience the cutaneous symptoms without weakness (dermatomyositis sine myositis), and 10% have muscle weakness without cutaneous symptoms. Initial cutaneous symptoms often include burning and pruritus, which may be associated with exposure to ultraviolet light or sunlight. Muscle weakness is characterized predominantly by proximal hip and shoulder muscle involvement: patients may have complaints of difficulty standing from a sitting position or raising their arms above their heads. They also have complaints of pain and tenderness to their muscles.1
Laboratory tests to support the diagnosis of dermatomyositis include serum muscle enzyme concentrations as well as autoantibody tests. Often, CK, LDH, aldolase, and aminotransferases are elevated from muscle breakdown. Patients usually have autoantibodies ranging from nonspecific antinuclear antibodies to the more specific anti–155 kDa. Electromyography (EMG) is characterized by increased irritability with spontaneous fibrillation and sharp waves. Often, skin and muscle biopsies show inflammatory changes, segmental necrosis, or other nonspecific findings. The diagnosis is confirmed through clinical history and examination of proximal muscle weakness with skin findings and 2 of 3 laboratory criteria. These include elevated muscle enzymes, EMG changes, and tissue biopsy, as described above.1
Although this patient had the typical findings of dermatomyositis, with confluent photosensitive rash over the malar area of his face and proximal muscle weakness, he also suffered from extensive calcinosis. While described as a complication of dermatomyositis in pediatric and adolescent patients, calcinosis is much less common in adult patients. Among the few cases seen in adults, calcinosis is often located in hard deposits around areas that experience frequent trauma (elbows and fingers).2 Socioeconomic status may play a role in the progression of calcinosis, as demonstrated in case reports3; this patient is a migrant worker without insurance. He demonstrated problems with timely follow-up to ensure he was getting the appropriate medicines after his initial diagnosis of rhabdomyolysis.
This patient developed extensive calcifications in the subcutaneous tissue of his right flank (Figure 1) and abdomen (Figure 2). This area is not typically affected in adults; however, the trauma from his appendectomy may have initiated an inflammatory response in this region. This inflammation likely precipitated the formation of the calcium deposits in his abdominal wall with resultant calcinosis.4
Treatment is largely based on controlling the likely autoimmune component of the disease. One possible etiology is complement-mediated inflammation at the vascular level; another is a direct cytotoxic effect of lymphocytes on the muscle cells. Initial therapy consists of high-dose steroids. Immunosuppressant and cytotoxic agents are often given early in the course in order to wean off steroids, thereby limiting the toxic effect of chronic steroids. Methotrexate, azathioprine, and mycophenolate mofetil are common agents used in dermatomyositis. If this combination of drugs is unsuccessful, intravenous immunoglobulins have shown promise for short-term treatment.5
Calcinosis is a difficult complication to treat. Some studies have shown success with diltiazem, aluminum hydroxide, and even alendronate in children.6,7 However, refractory cases of calcinosis that cause pain or interfere with function may need to be referred for surgical excision.8
Treatments for the rash are first focused on controlling systemic processes, but providing protection is also extremely important through limiting sun exposure and using sun protective clothing and sunscreen.
Current theories indicate calcinosis may be a consequence of untreated or unaggressively treated dermatomyositis. In juvenile dermatomyositis, early aggressive intervention offers the best protection from development of calcinosis. This adult patient had been experiencing symptoms for about 14 months before he received aggressive treatment, most likely another factor in his development of calcinosis.3 This patient suffered to the point at which he was no longer able to complete his activities of daily living. Luckily, he showed rapid signs of improvement with high-dose steroids and azathioprine. He was given prednisone and azathioprine also as an outpatient with continued improvement in his symptoms. Initially, intravenous immunoglobulin was considered because of the severity and progressive nature of his symptoms; however, it was never given owing to his response to other medicines.
The overall prognosis for patients treated with dermatomyositis is good, with a 5-year survival rate of 95% and a 10-year survival rate of 84%. Those who die from the condition often have pulmonary or cardiac manifestations as well. While most persons with dermatomyositis improve and respond to therapy, up to 30% experience long-term consequences.9 At a 2-month follow-up appointment, the patient was ambulating without assistance and was able to complete activities of daily living. He still reports some difficulty in standing from low sitting positions but continues to improve as his disease process is better controlled. The patient continues to take immunosuppressants and diltiazem. His calcifications remain, but his pain and symptoms are controlled.
Footnotes
Supervising Section Editor: Rick A. McPheeters, DO
Submission history: Submitted June 9, 2011; Revision received July 29, 2011; Accepted August 1, 2011
Reprints available through open access at http://escholarship.org/uc/uciem_westjem
DOI: 10.5811/westjem.2011.8.6823
Address for Correspondence: Bradley C. Presley, MD
Medical University of South Carolina, Division of Emergency Medicine, 169 Ashley Ave, MSC 300, Charleston, SC 29425
E-mail: brad.presley@gmail.com
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding, sources, and financial or management relationships that could be perceived as potential sources of bias. The authors disclosed none.
REFERENCES
1. Sontheimer RD, Costner MI. Wolff K, Goldsmith LA, Katz SI, editors. Dermatomyositis.Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill Professional; 2007.
2. Jorizzo J. Chapter 44: Dermatomyositis. In: Bolognia J, Jorizzo J, Rapini R, editors. Dermatology. 1st ed. Philadelphia, PA: Mosby;; 2003. pp. 115–122.
3. Weinel S, Callen J. Calcinosis cutis complicating adult-onset dermatomyositis. Arch Dermat.2004;140:365–366. [PubMed]
4. Redmond W, Baker S. Keloidal calcification. Arch Dermatol. 1983;119:270–272. [PubMed]
5. Dalakas M, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–982. [PubMed]
6. Mukamel M, Horev G, Mimouni M. New insight into calcinosis of juvenile dermatomyositis: a study of composition and treatment. J Pediatr. 2001;138:763–766. [PubMed]
7. Nakagawa T, Takaiwa T. Calcinosis cutis in juvenile dermatomyositis responsive to aluminum hydroxide treatment. J Dermatol. 1993;20:558–560. [PubMed]
8. Lobo I, Machado S, Teixeira M, et al. Calcinosis cutis: a rare feature of adult dermatomyositis.Dermatol Online J. 2008;14:10. [PubMed]
9. Dalakas MC. Fauci AS, Braunwald E, Kasper DL, Hauser SL, editors. Polymyositits, dermatomyositis, and inclusion body myositis. Harrison’s Principles of Internal Medicine. 17th ed.New York, NY: McGraw-Hill Professional; 2008.