
{kind=link}
| Author | Affiliation |
|---|---|
| Allan R Mottram, MD | University of Wisconsin School of Medicine and Public Health, Department of Medicine, Madison, Wisconsin |
| Sean M Bryant, MD | Cook County-Stroger Hospital, Department of Emergency Medicine, Chicago, Illinois |
| Steven E Aks, DO | Cook County-Stroger Hospital, Department of Emergency Medicine, Chicago, Illinois |
ABSTRACT
Introduction:
Sulfobutylether-β-cyclodextrin (SBE-CD) is a pharmaceutical excipient known to bind verapamil. Following intravenous administration, clearance of SBE-CD approximates glomerular filtration rate. We hypothesized that infusion of SBE-CD would increase time to asystole in a rat model of verapamil toxicity in a dose-dependent manner. The objective was to demonstrate the effect of a range of SBE-CD concentrations in a rat model of verapamil toxicity.
Methods:
Twenty-five Wistar rats were allocated to control or 1 of 4 intervention groups. All received ketamine and diazepam anesthesia followed by verapamil infusion 32 mg/kg/h. The verapamil infusion for the intervention groups was premixed with SBE-CD in a 1:1, 1:2, 1:4, or 1:8 molar ratio (verapamil to SBE-CD). The control group infusion did not contain SBE-CD. Additional saline or water was added to the infusion so that the total volume infused was the same across groups, and the osmolality was maintained as close to physiologic as possible. Heart rate, respiratory rate, and temperature were monitored. The primary endpoint was time to asystole.
Results:
Verapamil coinfused with SBE-CD in a molar ratio of 1:4 resulted in prolonged time to asystole compared to control (21.2 minutes vs 17.6 minutes, P < 0.05). There were no differences in time to asystole between control and any other intervention group. There was no significant difference in time to apnea between control and any intervention group. We assessed the effect of a range of SBE-CD concentrations and identified 1 concentration that prolonged time to asystole. Mechanisms that may explain this effect include optimal volume expansion with a hyperosmolar cyclodextrin containing solution, complexation of verapamil within the hydrophobic cyclodextrin pore, and/or complexation within micelle-like aggregates of cyclodextrin. However, mechanistic explanations for the observed findings are speculative at this point.
Conclusion:
The 1:4 verapamil to SBE-CD concentration was modestly effective with SBE-CD concentrations above and below this range demonstrating nonstatistically significant improvements in time to asystole.
INTRODUCTION
Cyclodextrins are hydrophilic circular oligosaccharides of various sizes containing a hydrophobic core. Lipophilic molecules fit into this hydrophobic core via nonionic bonds. Altering the number of substituent groups to the outer ring of the molecule significantly changes its characteristics, including its affinity for complexing with drugs and its osmolarity. They are widely used as pharmaceutical excipients to modify drug solubility and stability.1
Rather than improving drug delivery, we are interested in the ability of cyclodextrins to enhance elimination of drug from the body. This concept has proven feasible by the success of sugammadex, a gamma-cyclodextrin that was modified to function as an intravenous reversal agent for rocuronium-induced neuromuscular blockade following anesthesia.2,3 Cyclodextrin molecules bind target drugs as a function of a complexation constant.4 Modification of hydroxyl groups at the outer ring of the hydrophobic core enhances this property. We hypothesize that a favorable complexation constant, in addition to an equilibrium inequality driving the formula towards complexation, would allow binding and subsequent renal elimination of the target drug.
Additional mechanisms may contribute to reversal of toxicity. Micelle-like aggregates of cyclodextrin and drug-cyclodextrin complexes may solublize lipophilic drugs.5 By creating an intravascular sink, the drug is prevented from reaching the target organ. This is analogous to one proposed mechanism for how intralipid infusion works in the setting of bupivicaine, chlomipramine, and verapamil overdose.6–8
The study of cyclodextrins as antidotal therapy does appear in the literature. However, in contrast to the massive body of work regarding pharmaceutical applications of cyclodextrins, those regarding toxicologic applications are minimal. In-vitro inactivation of sarin and soman, treatment of organophosphate poisoning, and treatment of tunicaminyluracil toxicity is reported.9–12 The utility of cycodextrins as antidotes for the most common cardiovascular and neurologic toxins has not been evaluated.
The mechanism of drug reversal with intravenous cyclodextrin infusion is sound and has proven efficacy (ie, sugammadex). As such, we sought to apply this concept to models of drug toxicity which have shown significant morbidity and mortality and for which there are currently limited therapeutic options. Potential drug candidates that fit these criteria included tricyclic antidepressants, propoxyphene, calcium-channel antagonists, methamphetamine, and cocaine among others. Selecting the ideal drug was aided by the pharmaceutical chemistry literature. Verapamil is known to complex well with sulfobutylether-β-cyclodextrin (SBE-CD) as evidenced by bench work of verapamil enantiomeric separation.13,14
We have previously undertaken a study assessing the utility of SBE-CD as antidotal therapy for verapamil-induced cardiotoxicity in rats.15 This was a rescue model that used the same verapamil infusion as in the current study but a much higher concentration of SBE-CD (1:16 molar ratio of verapamil to cyclodextrin; 2.25 g/kg SBE-CD, 32 mg/kg/h verapamil). This was administered as a bolus after the onset of cardiotoxicity. Survival times were poorer in the cyclodextrin group, which we proposed was primarily related to the hyperosmolar load that accompanied the SBE-CD infusion (1,025 mOsm/kg) in combination with verapamil-induced cardiogenic shock. Additional confounding factors included use of a rescue model that induced severe, refractory cardiogenic shock prior to administration of our study drug, and the occurrence of isoflurane induced apnea. As such, with the current study, we used isoflurane for induction only, maintained anesthesia with ketamine and diazepam, studied a range of SBE-CD doses, and designed the study as a coinfusion model rather than a rescue treatment model. The aim of this study was to demonstrate the effect of a range of SBE-CD concentrations in a rat model of verapamil toxicity.
METHODS
The study design was an unblinded controlled trial to assess the effect of SBE-CD infusion in a rat model of verapamil toxicity. The protocol was approved by the Animal Care Committee at the University of Illinois Chicago. Male Wistar rats (strain 003) weighing 224 to 301 g with indwelling central venous femoral lines were purchased from Charles River Laboratories and housed in single cages with free access to food and water. Verapamil (2.5 mg/mL) was purchased from Hospira Inc (Lake Forest, Illinois). A 30% w/v solution of SBE-CD was prepared by dissolving 3 g dry SBE-CD in 100 mL of sterile water.
Preceding the experimental protocol, rats were allocated to either the control group (n = 5) or 1 of 4 intervention groups (n = 20). All rats underwent induction with 3% isoflurane via enclosed box. They were then weighed, and anesthesia was maintained with weight-based doses of ketamine (90 mg/kg initial; 30 mg/kg supplemental) and diazepam (6 mg/kg initial; 2 mg/kg supplemental). Following this, they were placed on a warming pad, and central venous lines were accessed for administration of verapamil and SBE-CD via infusion pump (Outlook 100 B.Braun, Bethlehem, Pennsylvania). Cardiac monitoring electrodes were attached using alligator clips for continuous monitoring (Escort II, Medical Data Electronics, Arleta, California). The study protocol was initiated with 5 minutes of observation under anesthesia for all rats to ensure stable respiratory rate and heart rate while maintaining adequate depth of anesthesia. Following this, the verapamil and SBE-CD infusion was initiated via central line. Verapamil (32 mg/kg/h) and SBE-CD (dose specified later) were compounded into the same infusion bag. The drugs were diluted in normal saline with additional saline and/or water added to the final concentrations as needed such that each subject would receive the same volume infusion (total volume 18.6 ml/kg/h across all groups) and the maximum osmolality would be limited. The verapamil dose was selected as it is the established LD50. It was efficacious in our prior work and is only slightly less than the dose established by Tebbutt et al (37.5 mg/kg/h) in a similar rat model of verapamil toxicity.15,7 The control group (group 1, n = 5) received no SBE-CD. The 4 intervention groups received verapamil and SBE-CD with the SBE-CD dose being 141 mg/kg (group 2, n = 5, 1:1 molar ratio of verapamil to SBE-CD), 282 mg/kg (group 3, n = 5, 1:2 molar ratio of verapamil to SBE-CD), 564 mg/kg (group 4, n = 5, 1:4 molar ratio of verapamil to SBE-CD), and 1,227 mg/kg (group 5, n = 5, 1:8 molar ratio of verapamil to SBE-CD). Infusate osmolality varied between 300 mOsm/L (1:1 infusion) and 352 mOsm/L (1:8 infusion). Of note, SBE-CD is specified to have no effect when administered to rats intravenously up to 2,000 mg/kg as documented in commonly available materials safety data sheet documents. Heart rate, respiratory rate, and temperature were monitored from onset of anesthesia to asystole or 1 h. The primary endpoint was time to asystole. Statistical analysis was performed using SPSS Statistics 18.0 (SPSS Inc, Chicago, Illinois). Data were analyzed for descriptive characteristics to ensure it was normally distributed. Further analysis was via paired t tests (discrete data) or one-way analysis of variance with Bonferroni post-hoc t tests (continuous data) as appropriate. Significance was set at P < 0.05. Figures were generated via Origin 7.5 (OriginLab Corporation, Northampton, Massachusetts).
RESULTS
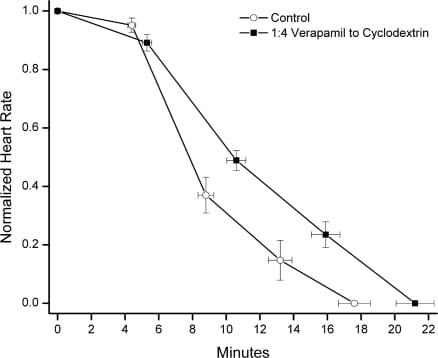
The baseline heart rate did not vary between control and intervention groups over the first 5 minutes of anesthesia, prior to initiation of verapamil and SBE-CD infusion. The baseline respiratory rate did vary between control and intervention groups 2 (mean difference 18.2, 95% confidence interval [CI] 4.7–31.7, P < 0.05) and 4 (mean difference 21.5, 95% CI 7.9–35, P < 0.05) during this time period (Figure 1). There were no significant differences between control and intervention group heart rate or respiratory rate during the drug infusion period (Figure 1). Time to asystole was significantly prolonged in group 4 compared to control (21.2 vs 17.6 minutes, P < 0.05; Figure 2). There were no other differences in time to asystole between control and intervention groups (Figure 1). There was no significant difference in time to apnea between control and any intervention group (Figure 1).

DISCUSSION
This study sought to identify a treatment benefit defined as a prolonged time to asystole in verapamil cardiotoxic rats coinfused with 141 mg/kg, 282 mg/kg, 564 mg/kg, or 1,227 mg/kg SBE-CD. These doses corresponded to a 1:1, 1:2, 1:4, and 1:8 molar ratio of verapamil to cyclodextrin respectively. The group infused with verapamil and SBE-CD in a 1:4 ratio demonstrated prolonged time to asystole. It is possible that this resulted from complexation of SBE-CD with verapamil, either in the infusion bag or intravascularly, followed by increased renal clearance, decreased tissue concentration, and reduced toxicity. Complexation and sequestration of verapamil within the hydrophobic cyclodextrin pore or within micelle-like aggregates of cyclodextrin is an intriguing mechanism, though speculative at this point. An alternative explanation is that 18 ml/kg/h of hypertonic solution (315 mOsm/kg) provided an ideal intravascular volume expansion, thus prolonging time to asystole. Arguing against this alternative is the fact that the solution administered to the 1:2 group was of the same approximate osmolality (318 mOsm/kg) and did not show any improvement in time to asystole.
LIMITATIONS
This project has several significant limitations. Rats in all groups experienced apnea very early in the course of the protocol, despite the modification of our protocol (ie, the use of diazepam and ketamine). Ideally, they would have been artificially ventilated to remove apnea as a confounding variable. We were unable to perform invasive hemodynamic monitoring. Such a capability would have allowed us to better characterize the hemodynamic effects of SBE-CD. Lastly, a small n, utilizing a small animal model, and lack of clear mechanism of action limits the applicability of our findings.
CONCLUSION
In conclusion, this study explores the application of a novel molecule to a challenging poisoning scenario. It demonstrated prolonged time to asystole in rats infused with both verapamil and SBE-CD compared to those infused with verapamil only. While acknowledging its limitations, the study provides support for further investigation of the use of cyclodextrins as antidotes to drug toxicity.
Footnotes
Supervising Section Editor: Brandon K. Wills, DO, MS
Submission history: Submitted December 30, 2010; Revision received March 2, 2011; Accepted March 21, 2011
Reprints available through open access at http://escholarship.org/uc/uciem_westjem
DOI: 10.5811/westjem.2011.3.6696
Address for Correspondence: Allan R. Mottram, MD
University of Wisconsin School of Medicine and Public Health, Department of Emergency Medicine, F2/204 Clinical Science Center MC3280, 600 Highland Ave, Madison, WI 53792
E-mail: allan.mottram@gmail.com
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding, sources, and financial or management relationships that could be perceived as potential sources of bias. The authors disclosed none.
REFERENCES
1. Challa R, Ahuja A, Ali J, et al. Cyclodextrins in drug delivery: an updated review. AAPS PharmSciTech. 2005;6:E329–E357. [PMC free article] [PubMed]
2. Bom A, Bradley M, Cameron K, et al. A novel concept of reversing neuromuscular block: chemical encapsulation of rocuronium bromide by a cyclodextrin-based synthetic host. Angew Chem Int Ed Engl. 2002;41:266–270. [PubMed]
3. Sparr HJ, Vermeyen KM, Beaufort AM, et al. Early reversal of profound rocuronium-induced neuromuscular blockade by sugammadex in a randomized multicenter study: efficacy, safety, and pharmacokinetics. Anesthesiology. 2007;106:935–943. [PubMed]
4. Szejtli J. Comprehensive Supramolecular Chemistry. New York, NY: Elsevier;; 1996.
5. Loftsson T, Magnusdottir A, Masson M, et al. Self-association and cyclodextrin solubilization of drugs. J Pharm Sci. 2002;91:2307–2316. [PubMed]
6. Weinberg GL, VadeBoncouer T, Ramaraju GA, et al. Pretreatment or resuscitation with a lipid infusion shifts the dose-response to bupivacaine-induced asystole in rats. Anesthesiology.1998;88:1071–1075. [PubMed]
7. Tebbutt S, Harvey M, Nicholson T, et al. Intralipid prolongs survival in a rat model of verapamil toxicity. Acad Emerg Med. 2006;13:134–139. [PubMed]
8. Harvey M, Cave G. Intralipid outperforms sodium bicarbonate in a rabbit model of clomipramine toxicity. Ann Emerg Med. 2007;49:178–185. [PubMed]
9. Desire B, Saint-Andre S. Interaction of soman with beta-cyclodextrin. Fundam Appl Toxicol.1986;7:646–657. [PubMed]
10. Desire B, Saint-Andre S. Inactivation of sarin and soman by cyclodextrins in vitro. Experientia.1987;43:395–397. [PubMed]
11. Verster RS, Botha CJ. Evaluation of hydroxypropyl-beta-cyclodextrin in the treatment of aldicarb poisoning in rats. J S Afr Vet Assoc. 2004;75:182–185. [PubMed]
12. May C, Stewart PL. Development of a toxin-binding agent as a treatment for tunicaminyluracil toxicity: protection against tunicamycin poisoning of sheep. Aust Vet J. 1998;76:752–756.[PubMed]
13. Xie GH, Skanchy DJ, Stobaugh JF. Chiral separations of enantiomeric pharmaceuticals by capillary electrophoresis using sulphobutyl ether beta-cyclodextrin as isomer selector. Biomed Chromatogr. 1997;11:193–199. [PubMed]
14. Chankvetadze B, Burjanadze N, Pintore G, et al. Chiral recognition of verapamil by cyclodextrins studied with capillary electrophoresis, NMR spectroscopy, and electrospray ionization mass spectrometry. Chirality. 1999;11:635–644. [PubMed]
15. Mottram AR, Aks SE, Bryant SM. Effect of cyclodextrin infusion in a rat model of verapamil toxicity. Am J Ther. 2011;18:371–374. [PubMed]