
{kind=link}
| Author | Affiliation |
|---|---|
| Jeffrey R. Suchard, MD | Department of Emergency Medicine, University of California, Irvine School of Medicine |
| Thomas A. Grotsky, MD | Department of Emergency Medicine, University of California, Irvine School of Medicine |
ABSTRACT
A 29-year-old man with no history of diabetes ingested over 60 grams of metformin in a suicide attempt. He presented to the emergency department with acute renal insufficiency, severe lactic acidosis, and rapidly-progressive hyperglycemia. The patient’s peak serum glucose level of 707 mg/dL is the highest yet reported in a case of metformin toxicity. Treatment included sodium bicarbonate infusion and hemodialysis, but the patient suffered several cardiac arrests with pulseless electrical activity and ultimately expired 25 hours after the ingestion.
INTRODUCTION
Metformin is a biguanide anti-hyperglycemic drug which is the most commonly prescribed oral agent to treat diabetes mellitus.1 Metformin-associated lactic acidosis (MALA) occurs infrequently with therapeutic use, occurring in 0.03 cases per 1000 patient-years.2 This rate is substantially lower than the incidence of phenformin-associated lactic acidosis, which caused that biguanide drug to be withdrawn from the U.S. market in the late 1970s.2–4 In overdose situations, however, metformin is frequently associated with lactic acidosis.3,5–15
Because the biguanide drugs do not enhance insulin release, as occurs with the sulfonylurea and meglitinide classes of diabetes medications, disorders of glucose homeostasis are rare with metformin. Although hypoglycemia associated with metformin has been reported, it is uncommon and often ascribed to concurrent use of other diabetes drugs.3,14,16 Hyperglycemia associated with metformin toxicity is even more rare, and has previously been ascribed to the patients’ underlying diabetes or to acute pancreatitis.3,14,17,18 We report the case of an intentional overdose of metformin in a patient without diabetes which resulted in progressive hyperglycemia early in the clinical course and fatal lactic acidosis. This patient’s peak serum glucose level of 707 mg/dL is the highest reported in a case of metformin toxicity.
CASE REPORT
A 29-year-old man ingested metformin in a suicide attempt. The patient consumed the entire remaining contents of his father’s prescription metformin bottle that originally contained 100 tablets of 850 mg each. The father stated that the bottle had contained at least three-quarters of its original contents, putting the ingested dose between 64 and 85 grams. The patient also consumed ethanol, but denied any other co-ingestants. The parents discovered the overdose around 6:30 a.m., about 5 ½ hours post-ingestion, when the patient began complaining of vomiting, diarrhea, thirst, abdominal pain and bilateral leg pain. Paramedics were called, who found the patient to be agitated with a fingerstick glucose level of 180 mg/dL.
The patient had a history of psychosis and depression, including prior suicide attempts by drug ingestion. He was not taking any prescribed medications, having discontinued olanzapine and sertraline several months earlier. The patient had no personal history of diabetes, despite the family history of type II diabetes in his father, who was taking no other anti-diabetic medications than metformin. The patient admitted to daily ethanol and tobacco use, but denied any current or past use of illicit drugs. He had no surgical history or known allergies.
Vital signs on arrival to the Emergency Department (ED) were temperature of 35.2°C (rectal), pulse of 113 beats/min, blood pressure of 129/59 mmHg, respirations at 28 breaths/min with 100% saturation via pulse oximetry on room air. The patient was awake and oriented x4, but agitated and slightly confused (GCS=14). Pupils were equal and reactive at 4mm and the oral mucous membranes were dry. Other than tachycardia, the heart and lung exams were unremarkable. The abdomen was mildly tender to palpation diffusely, but soft and without guarding or rebound. The patient had been incontinent of feces prior to arrival, but the stool was guaiac-negative and the patient had normal rectal tone. Electrocardiographic monitoring demonstrated a narrow-complex sinus tachycardia. A repeat fingerstick glucose upon arrival to the ED was 364 mg/dL. The patient became increasingly combative, attempting to bite and spit at the ED staff. He was placed in 4-point soft restraints, had a respiratory protection mask placed over his mouth, and was medicated with intravenous lorazepam 2 mg, dolasetron 12.5 mg, morphine sulfate 4 mg, and hydration with 0.9% saline. Within 45 minutes, the patient was no longer combative, and the physical restraints were removed.
Laboratory evaluation from blood samples obtained about one-half hour after ED arrival are shown in Table 1. Notably, the serum glucose level was 707 mg/dL associated with an elevated anion-gap metabolic acidosis. Arterial blood gas analysis showed: pH 7.10, pCO218.8 mmHg, and pO2 133.1 mmHg. The serum lactate level was above our clinical laboratory’s reporting range (>11.1 mmol/L). Even accounting for ethanol’s contribution to total osmolality, the calculated osmolal gap was highly elevated at >20 mOsm/kg. Given the presence of hyperglycemia with severe metabolic acidosis, the possibility of diabetic ketoacidosis was briefly considered; however, the serum β-hydroxybutyrate level was within normal limits (2.4 mg/dL [0.5–3.0]) and ketonuria was absent. Urinalysis showed glycosuria and moderate hemoglobin, but no calcium oxalate crystalluria, blood cells, or signs of infection. A urine drug screen by enzyme-multiplied immunoassay technique was negative for opiates, cocaine, phencyclidine, amphetamines, propoxyphene, benzodiazepines, barbiturates, and methylenedioxymethamphetamine.
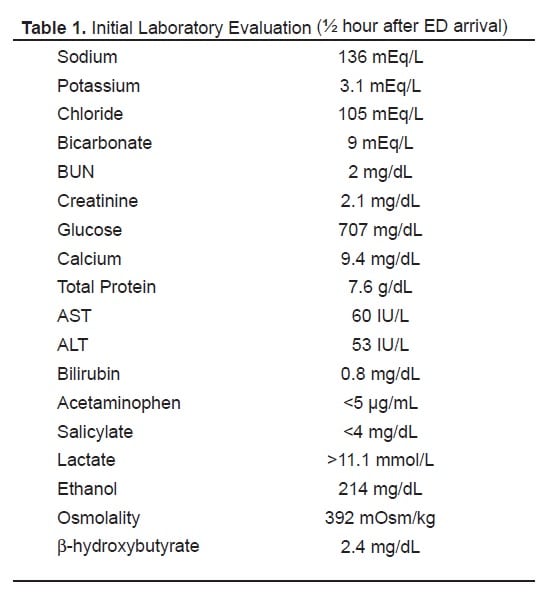
Initial Laboratory Evaluation (½ hour after ED arrival)
The patient was admitted to the medical intensive care unit service. Due to lack of ICU bed availability, the patient physically remained in the ED for several more hours. The nephrology service was initially contacted one hour after the patient’s arrival for consideration of emergent hemodialysis.
Repeat laboratory analysis on serum samples obtained four hours after ED arrival showed decreasing bicarbonate (6 mEq/L) and continued severe hyperglycemia (672 mg/dL), despite continued IV hydration with 0.9% saline boluses. The nephrology fellow was at the bedside discussing the benefits and risks of dialysis, when the patient was witnessed to become unresponsive, vomit, aspirate, and go into pulseless electrical activity (PEA) with a heart rate of 29/min. The patient was resuscitated with chest compressions, endotracheal intubation, 2 mg IV epinephrine, 1 mg IV atropine, and 4 ampules (176 mEq) of 8.4% sodium bicarbonate, resulting in a heart rate of 110/min and blood pressure 121/88 mmHg. An intra-resuscitative ABG showed: pH 6.95, pCO2 55.7 mmHg, pO288.1 mmHg, and bicarbonate 12.4 mEq/L. The patient was started on a bicarbonate drip (150 mEq NaHCO3 in 1L 5% dextrose at 200 cc/hr) and transferred to the ICU.
The patient developed non-sustained episodes of ventricular tachycardia, and then his blood pressure fell to 72/44 mmHg. A dopamine drip was started, but by the time the blood pressure stabilized the patient was found to be coagulopathic (INR 3.08; fibrinogen 113 mg/dL [156–400]; d-dimer >8.0 mcg/mL [<0.5]) and insertion of a hemodialysis catheter was delayed until the patient was treated with four units of fresh frozen plasma. The concurrent platelet count was 390 k/mm3 and the hematocrit was 47.7%. An echocardiogram showed mildly decreased left ventricular function, although serial cardiac enzymes showed progressive elevation of the CK-MB fraction and troponin I (up to 1.321 ng/mL [<0.03]). Hemodialysis was begun twelve hours after ED arrival. Three hours later, the patient again developed PEA and was resuscitated. Several more episodes of PEA recurred, and the patient expired about 19 hours after ED arrival. The last set of laboratory results, drawn five hours prior to death, showed a serum lactate level still >11.1 mEq/L, bicarbonate 12 mEq/L, and glucose 327 mg/dL. The hyperglycemia persisted despite treatment with an insulin drip (3 units regular insulin/hr) and the hemodialysis. No measurements of serum lipase or amylase were made during the patient’s hospitalization.
DISCUSSION
Overdoses with metformin are relatively uncommon, but may have serious consequences. In a five-year review of toxic exposures reported to U.S. poison control centers, only 4072 out of nearly 11 million exposures involved metformin, corresponding to less than one in 2500.3 There were a total of 9 deaths (0.2% of all metformin-related exposures), 32 cases with life-threatening signs or symptoms and/or residual disability (0.8%), and 187 cases with moderate clinical effects (4.6%). Cases of serious toxicity from metformin overdose are rare enough that single case reports and small case series continue to be published, which often describe extracorporeal methods of managing the consequent severe lactic acidosis.5–13,15
Lactic acidosis may occur either with therapeutic metformin dosing or in overdose; 0.03 cases of lactic acidosis per 1000 patient-years occur with therapeutic dosing, and most of these cases occur among patients who have contraindications to metformin, such as renal insufficiency.2 In overdose situations, lactic acidosis is seen much more commonly, although the exact incidence is unclear. Lactic acidosis was seen in 1.6% of metformin exposures reported to poison control centers, but only 10% of these exposures were due to intentional overdoses.3 In a review of poison control center inquiries from Germany, the incidence of metformin-associated lactic acidosis was 12.8% (14 of 109 calls); however, the incidence of attempted suicide was 56.9% in this group, suggesting that higher metformin doses were involved.11 Case reports and series of severe toxicity from metformin are typically associated with severe lactic acidosis,5–13,15,17,18 and many of these are due to intentional overdose.5–10,12,13,15,18 Such publications, however, form an obviously biased group, and the actual incidence of lactic acidosis with intentional metformin overdose remains unknown.
The prognosis in cases of undifferentiated lactic acidosis is poor, with an expected case-fatality rate of 30–50%.2,19 In cases of metformin-associated lactic acidosis, the serum lactate level does not correlate with prognosis, even with lactate levels as high as 35.5 mmol/L.19 In cases where metformin levels have been measured, there is a poorer prognosis with lower metformin levels.19 This unusual finding suggests that patients developing lactic acidosis at lower metformin levels probably have concurrent underlying disease, placing them at higher risk both for accumulating lactate and for death.
The most striking feature of the case reported here, however, is the profound and progressive hyperglycemia, which has not been previously reported. Metformin generally does not cause significant alterations in serum glucose levels, even in overdose situations. Unlike the sulfonylurea and meglitinide classes of diabetes drugs, which stimulate insulin release from the pancreas and therefore lower the glucose level, the biguanide drugs have more complex effects on glucose homeostasis that tend to reduce hyperglycemia without inducing hypoglycemia. These effects include: inhibition of hepatic gluconeogenesis (primarily through inhibiting hepatic lactate uptake), improving peripheral insulin sensitivity, inhibition of fatty acid oxidation, and possibly reducing intestinal glucose absorption.2,19 Metformin also increases production of lactate by the intestinal mucosa and suppresses pyruvate carboxylase, which impairs lactate clearance. Thus, lactic acidosis may occur in patients who have overdosed on metformin, or those with renal insufficiency, whose lactate clearance is already impaired.
Although metformin does not directly lower glucose levels, metformin-associated hypoglycemia has been reported, both with therapeutic dosing and with overdoses.3,14,16Hypoglycemia was reported in 2.8% of metformin exposures reported to a poison control center.3 In many of these cases the patients were receiving concurrent treatment with sulfonylurea drugs, which likely contributed in causing hypoglycemia. Some instances of drug interactions with metformin have been associated with hypoglycemia (e.g, with enalapril, which increases insulin sensitivity)20, although hypoglycemia with isolated metformin exposure is very rare.
Hyperglycemia associated with metformin overdose has occasionally been reported, but is even less common than hypoglycemia. In their review of 4072 metformin exposures, Spiller and Quadrani found only 18 cases associated with hyperglycemia (versus 112 with hypoglycemia), which they attributed as most likely due to the patients’ underlying diabetes.3 Nearly 40% of cases with hyperglycemia (seven of 18) occurred in patients who died, and seven of the nine overall fatalities had hyperglycemia, suggesting that it may be associated with particularly severe metformin toxicity.
Hyperglycemia has been related to acute pancreatitis in a few cases of metformin toxicity from both intentional overdose11,18 and therapeutic dosing.17 In the previously published cases of pancreatitis with metformin toxicity, the degree of hyperglycemia was considerably lower than in our patient: 162 mg/dL17, 345 mg/dL11, and 450 mg/dL (this final patient presented with hypoglycemia [23 mg/dL] and only became hyperglycemic after treatment with parenteral glucose).18 Similarly, our patient’s peak serum glucose exceeded the highest level (698 mg/dL) reported among diabetic patients taking metformin therapeutically who went on to develop lactic acidosis.21 Since our patient did not have a history of diabetes, his extreme hyperglycemia is all the more unexpected. Also, we could not identify any previously published cases where the hyperglycemia progressively worsened, in the absence of administration of glucose, as occurred in our patient early in his clinical course.
The potential mechanism for the severe hyperglycemia in our patient is not clear. Nothing among metformin’s known mechanisms would logically explain the progressive and severe hyperglycemia, especially since these mechanisms should tend to limit the glucose level. However, if one considers what might occur if the patient could no longer secrete enough insulin, as may occur with pancreatitis, then a potential explanation arises. With a lack of circulating insulin, metformin’s ability to enhance peripheral insulin sensitivity would count for nothing. Glucose would accumulate in the serum, since it would not be able to enter the tissues. It is also possible that a counter-regulatory hormone surge (epinephrine ± glucagon) from the acute physiologic stress of the overdose contributed to the hyperglycemia. This mechanism is conjectural, since pancreatitis was not confirmed in our patient with serum amylase and lipase levels, nor with any radiographic study showing pancreatic inflammation. Similarly, no circulating insulin levels were measured, which would have to be low for this mechanism to work. Nevertheless, pancreatitis remains a promising potential mechanism, as our patient’s clinical presentation with complaints of vomiting and abdominal pain is consistent with previously reported cases of metformin-associated pancreatitis.11,17,18
Alternate toxicologic causes for this patient’s hyperglycemia seem unlikely. Only a few toxins are routinely associated with hyperglycemia, including calcium channel-blockers and the ingestion of agents that specifically poison pancreatic beta-islet cells, such as alloxan, nitrophenolurea (an obsolete rodenticide), and streptozocin (a chemotherapy agent for pancreatic islet cell tumors). Calcium channel-blockers can impair insulin release, often resulting in modest hyperglycemia. Our patient had no known access to these drugs and did not present with hypotension or bradycardia. Similarly, our patient had no known access to beta-islet cell poisons, and the resultant hyperglycemia from these agents occurs in association diabetic ketoacidosis within a few days after significant exposure.
Our patient also ingested ethanol, which may have contributed to his severe toxicity. Ethanol itself may have increased the serum lactate level or induced pancreatitis. Also, the interaction of ethanol with biguanide drugs has been shown to increase lactate levels both in animal and human studies.4
Another potential complicating factor was the elevated osmolal gap in our patient, unaccounted for by the serum ethanol level. The presence of a highly elevated osmolal gap (>20 mOsm/kg) is most frequently caused by exposure to toxic alcohols,22 and our patient had a progressively worsening metabolic acidosis, which would be consistent with toxic alcohol poisoning. However, our patient denied ingesting anything but ethanol and metformin, and his parents confirmed that they saw no evidence at home of exposure to products containing isopropanol, methanol, or ethylene glycol. An unexplained osmolal gap may be seen in several severe disease states, including shock or sepsis,22 and was also seen in four out of nine cases of fatal metformin toxicity,3 providing an alternate explanation for its presence. We believe our patient’s severe acidosis and osmolal gap is highly unlikely to have occurred from toxic alcohol exposure for several reasons. Firstly, there was no historical evidence supporting the ingestion of any alcohol other than ethanol. Secondly, the patient did not exhibit additional clinical signs of end-organ damage, such as visual complaints from methanol toxicity, nor calcium oxalate crystalluria from ethylene glycol. The patient did have an elevated initial serum creatinine of 2.1 mg/dL, but this did not worsen as would be expected from ethylene glycol poisoning. Also, he had no ketonuria as would occur with isopropanol ingestion. Ingestion of propylene glycol might result in presentation with lactic acidosis and renal insufficiency, but such exposures are rare and severe acidosis should not occur in the presence of a supra-“therapeutic” ethanol level. Even if our patient had had severe toxic alcohol poisoning, the treatment would have included alcohol dehydrogenase inhibition, which was already effected by the presence of ethanol, and hemodialysis, which was already planned to treat his severe metformin toxicity.
CONCLUSION
Metformin toxicity is associated with development of lactic acidosis. Because metformin does not induce insulin release by pancreatic beta-islet cells, exposures infrequently induce disorders of glucose homeostasis. When abnormal glucose levels occur with metformin exposures, hypoglycemia is much more commonly seen than hyperglycemia; hypoglycemia is usually ascribed to concurrent anti-diabetic medications such as the sulfonylureas. Hyperglycemia appears to be a marker of severe toxicity in cases of metformin poisoning, and may be associated with the patient’s underlying diabetes or the development of pancreatitis.
We presented the case of a non-diabetic 29-year-old man who took a large overdose of metformin and ethanol in a suicide attempt. He presented with severe lactic acidosis and a progressively increasing serum glucose level. The hyperglycemia might have been due to pancreatitis, although confirmatory studies were not obtained before the patient died. The patient’s peak serum glucose level of 707 mg/dL is the highest yet reported in a case of metformin toxicity.
Footnotes
Supervising Section Editor: Brandon K. Wills, DO, MS
Submission history: Submitted February 19, 2008; Revision Received April 1, 2008; Accepted May 4, 2008.
Full text available through open access at http://escholarship.org/uc/uciem_westjem
Address for Correspondence: Jeffrey R. Suchard, MD. Department of Emergency Medicine, University of California, Irvine, 101 The City Drive, Rt. 128-01, Orange, CA 92868
Email: jsuchard@uci.edu
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding sources, and financial or management relationships that could be perceived as potential sources of bias. The authors disclosed none.
REFERENCES
1. Anonymous [Accessed February 12, 2008];Top 300 Prescriptions for 2005: By Number of US prescriptions dispensed. Available at: http://www.rxlist.com/script/main/art.asp?articlekey=79509.
2. Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. [PubMed]
3. Spiller HA, Quadrani DA. Toxic effects from metformin exposure. Ann Pharmacother.2004;38:776–780. [PubMed]
4. Chan NN, Brain HPS, Feher MD. Metformin-associated lactic acidosis: a rare or very rare clinical entity? Diabet Med. 1999;16:273–281. [PubMed]
5. Heaney D, Majid A, Junor B. Bicarbonate haemodialysis as a treatment of metformin overdose. Nephrol Dial Transplant. 1997;12:1046–1047. [PubMed]
6. Teale KFH, Devine A, Stewart H, Harper NJH. The management of metformin overdose. Anaesthesia. 1998;53:691–701. [PubMed]
7. Barrueto F, Meggs WJ, Barchman MJ. Clearance of metformin by hemofiltration in overdose. Clin Toxicol. 2002;40:177–180.
8. Chang CT, Chen YC, Fang JT, Huang CC. High anion gap metabolic acidosis in suicide: Don’t forget metformin intoxication – two patients’ experiences. Ren Fail. 2002;24:671–675. [PubMed]
9. Gjedde S, Christiansen A, Pedersen SB, Rungby J. Survival following a metformin overdose of 63 g: a case report. Pharm Toxicol. 2003;93:98–99.
10. Nisse P, Mathieu-Nolf M, Deveaux M, Forceville X, Combes A. A fatal case of metformin poisoning. J Toxicol Clin Toxicol. 2003;41:1035–1036. [PubMed]
11. von Mach MA, Sauer O, Weilemann LS. Experiences of a poison center with metformin-associated lactic acidosis. Exp Clin Endocrinol Diabetes. 2004;112:187–190.[PubMed]
12. Harvey B, Hickman C, Hinson G, Ralph T, Mayer A. Severe lactic acidosis complicating metformin overdose successfully treated with high-volume venovenous hemofiltration and aggressive alkalinization. Pediatr Crit Care Med. 2005;6:598–601.[PubMed]
13. Lacher M, Hermanns-Clausen M, Haeffner K, Brandis M, Pohl M. Severe metformin intoxication with lactic acidosis in an adolescent. Eur J Pediatr. 2005;164:362–365.[PubMed]
14. Spiller HA, Sawyer TS. Toxicology of oral antidiabetic medications. Am J Health-Syst Pharm. 2006;63:29–38.
15. Galea M, Jelacin N, Bramham K, White I. Severe lactic acidosis and rhabdomyolysis following metformin and ramipril overdose. Br J Anaesth. 2007;98:213–215. [PubMed]
16. Spiller HA, Weber JA, Winter ML, et al. Multicenter case series of pediatric metformin ingestion. Ann Pharmaother. 2000;34:1385–1388.
17. Ben MH, Thabet H, Zaghdoudi I, Amamou M. Metformin associated acute pancreatitis.Vet Human Toxicol. 2002;44:47–48.
18. Mallick S. Metformin induced acute pancreatitis precipitated by renal failure.Postgrad Med J. 2004;80:239–240. [PMC free article] [PubMed]
19. Lalau JD, Race JM. Lactic acidosis in metformin therapy. Drugs. 1999;58(Suppl 1):55–60. [PubMed]
20. Talwalkar PG, Deotale P. Severe hypoglycemia in a patient with type 2 diabetes mellitus on metformin monotherapy. J Assoc Phys India. 2007;55:458–459.
21. Lalau JD, Race JM. Lactic acidosis in metformin-treated patients: prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Safety. 1999;20:377–384. [PubMed]
22. Suchard J. Osmolal gap. In: Dart RD, et al., editors. Medical Toxicology. 3. Philadelphia: Lippincott Williams & Wilkins; 2004. pp. 106–109.