
{kind=link}
| Author | Affiliation |
|---|---|
| Charles R Wira, III, MD | Yale School of Medicine, Department of Emergency Medicine and Acute Stroke Service, New Haven, Connecticut |
| Emanuel Rivers, MD, MPH | Henry Ford Hospital, Department of Emergency Medicine and Surgical Critical Care, Detroit, Michigan |
| Cynthia Martinez-Capolino, MD | Henry Ford Hospital, Department of Emergency Medicine, Detroit, Michigan |
| Brian Silver, MD | Henry Ford Hospital, Department of Neurology, Detroit, Michigan |
| Gayathri Iyer, MD | Henry Ford Hospital, Department of Cardiology, Detroit, Michigan |
| Robert Sherwin, MD | Wayne State University, Department of Emergency Medicine, Detroit, Michigan |
| Christopher Lewandowski, MD | Henry Ford Hospital, Department of Emergency Medicine and Acute Stroke Service, Detroit, Michigan |
ABSTRACT
Introduction:
To characterize cardiac complications in acute ischemic stroke (AIS) patients admitted from an urban emergency department (ED).
Methods:
Retrospective cross-sectional study evaluating AIS patients admitted from the ED within 24 hours of symptom onset who also had an echocardiogram performed within 72 hours of admission.
Results:
Two hundred AIS patients were identified with an overall in-hospital mortality rate of 8% (n = 16). In our cohort, 57 (28.5%) of 200 had an ejection fraction less than 50%, 35 (20.4%) of 171 had ischemic changes on electrocardiogram (ECG), 18 (10.5%) of 171 presented in active atrial fibrillation, 21 (13.0%) of 161 had serum troponin elevation, and 2 (1.1%) of 184 survivors had potentially lethal arrhythmias on telemetry monitoring. Subgroup analysis revealed higher in-hospital mortality rates among those with systolic dysfunction (15.8% versus 4.9%; P = 0.0180), troponin elevation (38.1% versus 3.4%; P < 0.0001), atrial fibrillation on ECG (33.3% versus 3.8%; P = 0.0003), and ischemic changes on ECG (17.1% versus 6.1%; P = 0.0398) compared with those without.
Conclusion:
A proportion of AIS patients may have cardiac complications. Systolic dysfunction, troponin elevation, atrial fibrillation, or ischemic changes on ECG may be associated with higher in-hospital mortality rates. These findings support the adjunctive role of cardiac-monitoring strategies in the acute presentation of AIS.
INTRODUCTION
Acute central nervous system injury has long been associated with myocardial injury and dysfunction.1–3 Some have implicated serum catecholamine elevations in particular with strokes involving the insular cortex,4–6 but the mechanisms underlying this relation are poorly understood in acute ischemic stroke (AIS). Cardiac dysfunction in stroke patients may be particularly damaging. Within the territory of tissue affected by an AIS (ie, the ischemic penumbra) intrinsic autoregulation of the vasculature is lost, rendering cerebral blood flow directly dependent on cardiac function.7–9
However, a paucity of investigators have evaluated the incidence, role, and impact of cardiac function and complications with AIS. Emergency physicians responsible for managing AIS currently have no practice guidelines promoting cardiovascular monitoring beyond blood pressure control, continuous telemetry monitoring, cardiac biomarkers, and admission electrocardiograms (ECG).10Once admitted, AIS patients undergo inpatient echocardiograms evaluating for a cardioembolic source, with no recommendations regarding management of systolic or diastolic dysfunction. The identification of cardiac comorbidity is important, as future interventions such as cardiac augmentation may benefit a subset of stroke patients.11
We designed this preliminary study to reinforce the limited existing literature characterizing the prevalence of cardiac complications in the setting of AIS. For our primary outcome, we hypothesized that a significant proportion of AIS patients have systolic dysfunction on echocardiography. As alternative outcomes, we hypothesized that a significant proportion of AIS patients also have further myocardial or hemodynamic abnormalities manifested by ECG abnormalities, diastolic dysfunction on echocardiography, serum troponin elevation, elevated systolic blood pressure, or the presence of hemoconcentration, suggesting a low-volume state.
METHODS
This study was a single-reviewer retrospective cross-sectional analysis using defined markers to characterize the cardiac complications and the hemodynamic profile of AIS patients. Our primary outcome was the frequency of systolic dysfunction, defined as an ejection fraction (EF) on echocardiography less than 50%, as previously defined in the cardiology literature.12Echocardiograms were read by staff cardiologists at our hospital as a part of the standard care for all AIS patients.
Secondary outcomes were the presence of ECG abnormalities, diastolic dysfunction on echocardiography, serum troponin I elevation (greater than 0.04 mg/dL), an elevated initial systolic blood pressure, and the presence of a hemoconcentrated red blood cell volume. ECG abnormalities were originally read by a staff cardiologist at our hospital as a part of the formal medical record. The interpretations were then collected by a study investigator. An abnormal ECG included the presence of ischemic changes on ECG (ie, T-wave inversions, ST-segment changes) or the presence of arrhythmias (ie, atrial fibrillation). Diastolic dysfunction on echocardiography was measured in one of three ways: “E” (early atrial filling) to “A” (atrial kick) waveform reversal, tissue Doppler measurements on transthoracic echocardiography, or pulmonary vein flow on transesophageal echocardiography. Serum troponin values were included if the blood was drawn in the emergency department (ED) or within 24 hours of admission. An elevated initial systolic blood pressure was represented by the initial blood pressure measurement in the ED. The presence of hemoconcentration was measured by comparing the initial ED hemoglobin value to the mean of up to three prior outpatient serum hemoglobin measurements. Although this is a largely untested and indirect method for assessing fluid volume status, we assumed a low-fluid-volume state was present if the ED hemoglobin values were greater than the mean of three outpatient hemoglobin values.
The study was performed at an academic tertiary care hospital seeing more than 110,000 ED visits per year. The protocol and procedures were approved by the Institutional Review Board for human research. This study was a chart review of adult patients admitted from the ED over a 1-year period with the diagnosis of AIS. It was determined a priori that 200 patients would be included. Patients were identified from the ED electronic medical record, which tracked all admissions from the ED. Patients were identified from the electronic medical record for being admitted with a diagnosis of stroke, ischemic stroke, or cerebral vascular accident between January 1, 2004, and December 31, 2004. After a patient list was generated from the medical record, a senior resident investigator (C.W.) under the supervision and monitoring of senior faculty investigators (C.L. and E.R.) used a standardized data collection and glossary of terms to extract demographic and clinical data. The faculty mentors were involved in study design, Institutional Review Board approval, creation of the data-collection form, creation of a database, and were involved in the analysis of the data. Faculty mentors met with the resident on a regular basis to ensure compliance, to review questions and unforeseen issues related to data collection or abstraction, and were involved in the analysis ofdata.
Inclusion criteria were patients diagnosed with AIS in the ED who presented within 24 hours of onset of symptoms, and patients who had a cardiac echocardiogram performed within 72 hours of hospital admission. Exclusion criteria were age younger than 18 years, evidence of cerebral hemorrhage on initial head computed tomography, resolution of neurologic symptoms in the ED or ED diagnosis of transient ischemic attack (TIA), or the presence of documented chest pain in the ED.
For our primary analysis, sample size calculation, given that past literature13,14 demonstrated that 13% to 29% of AIS patients have cardiac dysfunction, we estimated sufficient power with a sample size of at least 200 AIS patient subjects to detect the prevalence of impaired contractility (EF less than 50%) within +5% of the true prevalence. Results were reported with the mean, standard deviation, and 95% confidence interval where indicated. Paired and unpaired t tests were used to compare means between different groups. The Fisher Exact Test was used in subgroup analysis for comparing in-hospital mortality rates among patients with systolic dysfunction, ECG abnormalities, or serum troponin I elevation. Statistical significance was indicated by an alpha error less than 0.05.
For our primary analysis, the established inclusion and exclusion criteria assured that no data were missing and that all patients had a measurement of contractility on echocardiography. For our secondary analyses, we aimed to estimate the prevalence of each abnormality (ie, troponin elevation), and to compare the difference in mortality rate contrasted to patients without the abnormalities (ie, patients with abnormal versus normal troponin levels). To estimate prevalence, we used the number of patients who actually had these tests performed as the denominator for calculating the prevalence of abnormal values (ie, the percentage of patients with abnormal ECGs). To compare differences in mortality rates, we first established as a prerequisite that at least 80% of patients would have the variable measured for each comparison. We then used the total patient sample (n = 200) as the denominator for comparing mortality rates by using the Fisher Exact Test between the different subgroups.
If other variables (ie, systolic blood pressure, National Institutes of Health stroke scale [NIHSS]) were not available in the ED medical record, then they would be acquired from the inpatient medical records, with the first value being reported being the one that was used. If the NIHSS was not available in the inpatient records, then it would be calculated retrospectively15 by the data extractor under the supervision of senior faculty. If a comprehensive admission neurologic examination was not documented in the medical record, then that patient was not included in analyses using the NIHSS. For the assessment of hemoconcentration, missing inpatient or outpatient hemoglobin values excluded patients from this alternative analysis.
RESULTS
In total, 398 patients with an ED admission diagnosis of stroke, ischemic stroke, or cerebral vascular accident were identified; 58 were excluded for having cerebral hemorrhage on initial imaging; and 140 were excluded for not meeting inclusion criteria, including being admitted with a primary diagnosis other than AIS or presenting with a duration of symptoms longer than 24 hours. Because our acute stroke protocol includes an echocardiogram on admission for all AIS patients, only 8 patients were excluded because echocardiograms were not performed within 72 hours of hospital admission. Cumulatively, 200 patients met inclusion criteria.
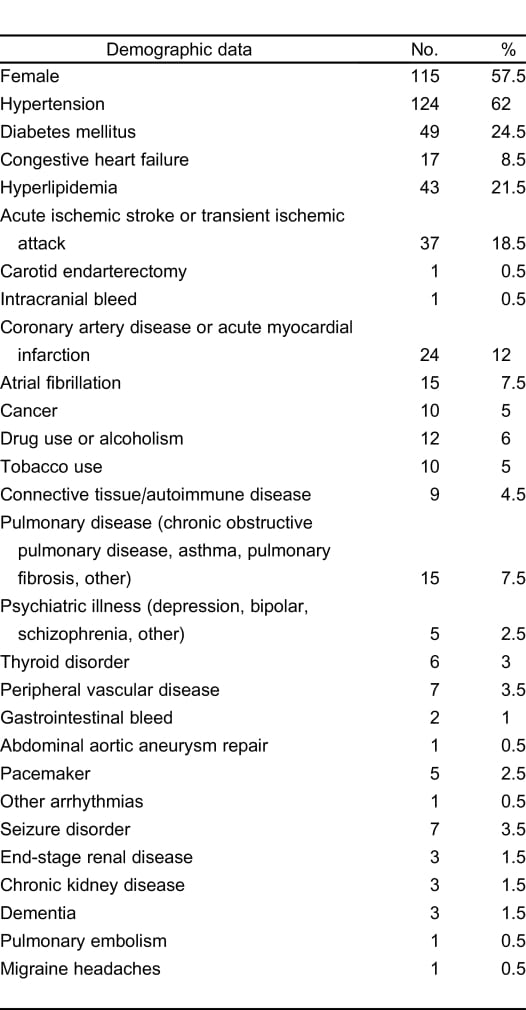
Demographics of our patient population are summarized in Table 1. Of our patients, 57.5% were women. The mean age was 65.2 + 14.9 years old. The most common comorbidities were hypertension (62%), diabetes (24.5%), and hyperlipidemia (21.5%). Of the patients, 18.5% previously had an AIS or TIA; 8.5% of patients had a history of congestive heart failure. For all patients, the mean initial systolic blood pressure for patients in the ED was 168.9 mm Hg + 37.8 (95% confidence interval [CI], 161.3 mm Hg to 176.5 mm Hg). Among all patients, the overall in-hospital mortality rate was 8% (n = 16). The mean initial NIHSS for identified patients was 7.88 + 6.15 (95% CI, 7.00 to 8.77; n = 188).
Of the total sample of patients, 57 (28.5%) had evidence of systolic dysfunction defined as an EF less than 50% on echocardiograms performed within 72 hours of hospital admission; 38 (18%) patients had evidence of diastolic dysfunction on echocardiograms, as indicated by E to A reversal, abnormal tissue Doppler, or abnormal pulmonary vein flow. Of the patients, 171 (85.5%) had ECGs performed in the ED; 35 (20.4%) patients had ischemic changes on ECG defined as T-wave inversions or ST-segment depressions. No patients had ST-segment elevations in concurrent ECG leads. Eighteen (10.5%) patients presented in active atrial fibrillation.
Among laboratory tests, 161 (80.5%) patients had serum troponin I levels checked in the ED or within 24 hours of hospitalization, with 21 (13.0%) having elevated values; 116 (58.0%) had outpatient hemoglobin values available for assessment of hemoconcentration, the surrogate marker for a low-volume state. Mean hemoglobin levels on initial presentation in the ED were higher contrasted to outpatient baseline values (13.5 + 1.7 g/L versus 12.6 + 1.5 g/L; n = 116, P = 0.0001 by using a paired t test).
Twenty-five patients received thrombolysis, with 19 having improvement of symptoms before hospital discharge. Among patients receiving TPA and having documented NIHSS in the ED and at discharge, the mean initial NIHSS in the ED was 12.8 + 9.82, and the mean predischarge NIHSS was 6.4 + 9.3 (P = 0.0775; n = 15). Two patients had symptomatic hemorrhagic transformation, defined as an escalation of NIHSS with evidence of cerebral hemorrhage on serial neuroimaging. One patient had asymptomatic hemorrhagic transformation.
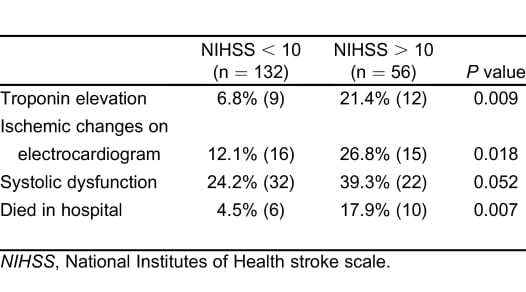
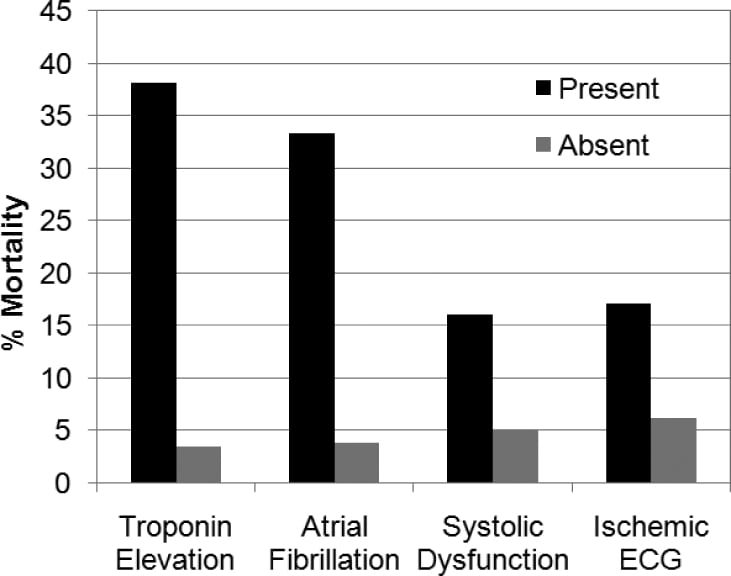
Patients with an NIHSS greater than 10, contrasted with those with an NIHSS of 10 or less, had higher rates of in-hospital mortality, serum troponin elevation, and ischemic changes on ECG. A trend existed toward a higher rate of systolic dysfunction among patients with more-severe strokes (Table 2). We did not observe mortality differences when evaluating blood pressure or for the presence of hemoconcentration. Subgroup analysis (Figure) with the Fisher Exact Test revealed higher in-hospital mortality rates among patients with systolic dysfunction (15.8% versus 4.9%; P = 0.0180), troponin I elevation (38.1% versus 3.4%, P < 0.0001), atrial fibrillation on initial ECG (33.3% versus 3.8%; P = 0.0003), and ischemic changes on initial ECG (17.1% versus 6.1%; P = 0.0398). Two survivors (1.1%; n = 184) had multiple (more than 3) episodes of nonsustained ventricular tachycardia documented on telemetry during hospitalization.
DISCUSSION
Heart–brain interactions are more comprehensively described by neurosurgical investigators in the setting of a specific type of hemorrhagic stroke, the acute subarachnoid hemorrhage (SAH). Indeed, in neurosurgical models and clinical studies, 40% to 100% with SAH can have ECG changes or arrhythmias,16–20 11% to 21% can have myocardial enzyme elevation,17,19,20 and 2% to 30% may have left ventricular dysfunction manifested by hypokinetic wall-motion abnormalities on echocardiography.17–20 Compared with AIS, heart–brain interactions in SAH have been more widely investigated,21–34 with literature describing reversible myocardial stunning,35 histologic evidence of myocardial necrosis,36 elevation of natriuretic factors,37,38 secondary neurogenic pulmonary edema,39 elevation of systemic plasma catecholamines,40,41 and impairment of regional myocardial perfusion.42,43 Additionally, in the neurosurgical literature, hemodynamic manipulations have been more rigorously evaluated, specifically for the management of cerebral vasospasm after SAH.44
In the setting of AIS, cardiac complications may be particularly damaging with the high incidence of underlying coronary artery disease,3,45 and given evidence that within the ischemic penumbra intrinsic autoregulation of the vasculature is often lost rendering cerebral blood flow directly dependent upon cardiac function.7,8 Our data compliments the limited existing literature performed by other investigators suggesting that 13% to 29%13,14 of AIS patients may have systolic dysfunction on echocardiography, 35% to 74%1–3,46 may have ischemic changes on ECG, and 1% to17%1,3,4,46–48may have elevated serum troponin levels, which has been shown to be associated with higher mortality rates.47,48
In our study, we used the indirect surrogate markers of initial systolic blood pressure and hemoconcentration to represent an estimate of afterload and preload, respectively. Although use of these surrogate markers is untested and affected by potential biases, our findings were compatible with existing literature. Current literature demonstrates that AIS patients may present with elevated afterload represented by an elevated mean arterial pressure.49 Only 1 study to our knowledge has evaluated preload, reporting a mean central venous pressure of 4.5 mm Hg.50 Additionally, even though this variable was not evaluated in our study, 2 studies to our knowledge have recorded cardiac output in a small sample of AIS patients.50,51
Regarding structural systolic function on echocardiography, we observed systolic dysfunction in 28.5% of patients, which is similar to that in other cited investigations.13,14 Although our design did not have a control group or allow follow-up echocardiography to determine whether this finding was only transient, only 8.5% of our patients had a pre-existing diagnosis of congestive heart failure. Thus it is possible that a large proportion of our patients may have had an acute change in cardiac contractility temporally related to their AIS (ie, either before or after). Nevertheless, similar to other investigators’ observations that undifferentiated systolic dysfunction is associated with impaired outcome,52 we also observed an association between systolic dysfunction and higher in-hospital mortality rates. Our preliminary results support the need for further investigation in this relatively unexplored area.
In a subgroup analysis of our study, we also observed an association between an elevated troponin or presenting in active atrial fibrillation and higher in-hospital mortality rates. This is consistent with prior literature. Barasch et al48 evaluated a small series of patients with troponin elevation who had no clinical manifestations of acute coronary syndromes, and concluded that troponin elevation did not predict hospital mortality. However, their subset of patients with ischemic and hemorrhagic strokes accounted for half of the in-hospital fatalities. James et al47 evaluated the relation between troponin T elevation and mortality in AIS patients. Their results identified mortality rates of 40% in patients with elevated troponin T compared with 13% in patients without elevations. Regarding atrial fibrillation, 1 study suggested that active atrial fibrillation appears to be a specific AIS predictor of in-hospital mortality.53 However, underlying mechanisms are not well understood.
Although evidence exists from the SAH literature and preliminary data in the AIS literature, limited investigation has evaluated prevalence and influence of cardiac complications and hemodynamic derangements in the setting of AIS. Furthermore, in AIS practice guidelines, no standardized recommendations promote hemodynamic monitoring beyond elementary blood pressure control, checking cardiac biomarkers, and telemetry monitoring. No recommendations exist for hemodynamic optimization or augmentation.10 Despite the paucity of data compared with those in the neurosurgical literature, our study complements existing studies, suggesting that AIS patients may acutely present with decreased contractility. Whether these findings occur as a consequence of the AIS, or whether they predispose individuals to have AIS, we believe this is an area that merits further investigation and that the data collected provide a good foundation for future research evaluating the underlying mechanisms, stroke subtypes at risk, and treatment alternatives for AIS patients manifesting cardiac complications.
What does this mean for the EMP managing these patients? Our results complement existing recommendations to perform ECGs, measure serum cardiac enzymes, and perform telemetry monitoring on all AIS patients in the acute presentation to the ED. Given in our study that only 85% (n = 171) of patients had ECGs performed in the ED, and that only 80% (n = 161) had cardiac enzymes drawn in the ED, this potentially is an important area for increased awareness, as cardiac complications may be associated with a worse outcome in patients with both large and small strokes. Furthermore, it is conceivable, given the evolution of the specialty of emergency medicine, that in the future, bedside ultrasound may be used noninvasively to estimate preload and evaluate for impaired contractility. Certainly our specialty literature has demonstrated that high-quality echocardiograms may be performed by EPs.54 Additionally, given the dependence of cerebral blood flow on cardiac function, it is plausible to envision the development of a stroke goal-directed hemodynamic optimization study aimed at linking systemic resuscitation in the acute presentation to the salvation of the penumbra.
In summary, although our study is limited by design, small sample size, potential bias, and lack of controls, it suggests that a significant proportion of ED AIS patients have cardiac complications that potentially may be associated with higher mortality rates. Our data also suggest that patients with more-severe strokes may be more likely to have associated cardiac complications. Thus, our results complement current guidelines,10 suggesting that cardiac biomarkers, an ECG, and telemetry monitoring be used in the ED assessment of all AIS patients. They also support the need for further investigation in the area of heart–brain interactions in the setting of AIS.
LIMITATIONS
Our study is limited by its retrospective design and small sample size. Additionally, we did not have a control group to identify whether a higher rate of systolic dysfunction occurred among AIS patients compared with age-matched controls. Nevertheless, the presence of almost a third of patients having undifferentiated systolic dysfunction—whether as an underlying comorbidity or acutely within the same period as the stroke—is a significant finding that merits further investigation. Limited studies complement our results by suggesting that undifferentiated systolic dysfunction is associated with worse functional neurologic outcome.52
Another limitation is that our method for identifying patients may have missed AIS patients admitted from the ED with another diagnosis (ie, altered mental status). However, this is a common limitation of stroke studies identifying patients in the ED.55 Additionally, because of the retrospective design of the study, a selection bias for the subgroup analyses involved serum troponin, ECG abnormalities, and red blood cell volume, as not all patients had these tests performed by treating physicians in the ED or outpatient setting.
Additionally, a limitation in our study was the use of a surrogate marker for volume status. Our method for evaluating hemoconcentration is untested and has potential bias. Even though we were not aware of any patients receiving blood transfusions in the ED or taking exogenous erythropoietin before presentation, which would have biased these results, we were unable to control for crystalloid volume administered before laboratory results were determined. However, we suspect that a large number of patients had blood drawn before crystalloid administration because in our institution’s acute stroke protocol, blood is typically drawn when the first intravenous injection is placed before the administration of any medications or fluids.
Given that echocardiography is a standard of care for all AIS patients in our institution to potentially identify a source of the stroke, we were reassured that only 8 patients were excluded for not having echocardiograms performed within 72 hours. However, a limitation of the retrospective nature of this study is that we were unable to control for concomitant medications or other variables that may have transiently affected systolic function when echocardiograms were being performed.
CONCLUSION
A proportion of ED AIS patients may have associated cardiac complications, either as a direct result of the AIS, or because of other factors that were not analyzed in this study. Serum troponin elevation, active atrial fibrillation, ischemic changes on ECG, and the presence of systolic dysfunction may be associated with more-severe strokes and with higher in-hospital mortality rates. These findings support the adjunctive role of cardiac-monitoring strategies in the acute presentation of AIS, and support the need for further research evaluating heart–brain interactions in the setting of AIS.
Footnotes
Supervising Section Editor: Matthew Strehlow, MD
Submission history: Submitted November 11, 2009; Revision received August 2, 2010; Accepted February 4, 2011
Reprints available through open access at http://escholarship.org/uc/uciem_westjem
DOI: 10.5811/westjem.2011.2.1765
Address for Correspondence: Charles R. Wira III, MD
Yale School of Medicine, Department of Emergency Medicine, 464 Congress Ave, Ste 260, New Haven, CT 06519-1315
E-mail: charles.wira@yale.edu
Conflicts of Interest: By the WestJEM article submission agreement, all authors are required to disclose all affiliations, funding sources, and financial or management relationships that could be perceived as potential sources of bias. The authors disclosed none.
REFERENCES
1. Oppenheimer SM, Hachinski VC. The cardiac consequences of stroke. Neurol Clin. 1992;;10:167–176. [PubMed]
2. Burch GE, Meyers R, Abildskov JA. A new electrocardiographic pattern observed in cerebrovascular accidents. Circulation. 1954;;9:719–723. [PubMed]
3. Norris JW, Hachinski VC, Myers J, et al. Serum cardiac enzymes in stroke. Stroke. 1979;;10:548–553. [PubMed]
4. Barber M, Morton JJ, Macfarlane PW, et al. Elevated troponin levels are associated with sympathoadrenal activation in acute ischaemic stroke. Cerebrovasc Dis. 2007;;23:260–266.[PubMed]
5. Sander D, Winbeck K. Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology. 2001;;57:833–838. [PubMed]
6. Meyer S, Strittmatter M. Lateralization in autonomic dysfunction in ischemic stroke involving the insular cortex. Neuroreport. 2004;;15:357–361. [PubMed]
7. Keller TS, McGillicuddy JE, LaBond VA, et al. Volume expansion in focal cerebral ischemia: the effect of cardiac output on local cerebral blood flow. Clin Neurosurg. 1982;;29:40–50. [PubMed]
8. Keller TS, McGillicuddy JE, LaBond VA, et al. Modification of focal cerebral ischemia by cardiac output augmentation. J Surg Res. 1985;;39:420–432. [PubMed]
9. Tranmer BI, Keller TS, Kindt GW, et al. Loss of cerebral regulation during cardiac output variations in focal cerebral ischemia. J Neurosurg. 1992;;77:253–259. [PubMed]
10. Adams HP Jr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the AHA/ASASC/CCC/CRIC. Stroke. 2007;;38:1655–1711.[PubMed]
11. Caplan L. Worsening in ischemic stroke patients: is it time for a new strategy? Stroke.2002;;33:1443. [PubMed]
12. Pfisterer M, Battler A, Zaret BL. Range of normal values for left and right ventricular ejection fraction at rest and during exercise assessed by radionuclide angiography. Eur Heart J. 1985;;6:647–655. [PubMed]
13. Rauh G, Fischereder M, Spengel FA. Transesophageal echocardiography in patients with focal cerebral ischemia of unknown cause. Stroke. 1996;;27:691–694. [PubMed]
14. Stouffer GA, Sheahan RG, Sorescu D, et al. Clinical and transthoracic echocardiographic predictors of abnormal transesophageal findings in patients with suspected cardiac source of embolism. Am J Med Sci. 2003;;326:31–34. [PubMed]
15. Williams LS, Yilmaz EY, Lopez-Yunez AM. Retrospective assessment of initial stroke severity with the NIH stroke scale. Stroke. 2000;;31:858–862. [PubMed]
16. Masuda T, Sato K, Yamamoto S, et al. Sympathetic nervous activity and myocardial damage immediately after subarachnoid hemorrhage in a unique animal model. Stroke. 2002;;33:1671–1676. [PubMed]
17. Elrifai AM, Bailes JE, Shoe-Ren S, et al. Characterization of the cardiac effects of acute subarachnoid hemorrhage in dogs. Stroke. 1996;;27:737–742. [PubMed]
18. Yoshikawa D, Hara T, Takahashi K, et al. An association between QTc prolongation and left ventricular hypokinesis during sequential episodes of subarachnoid hemorrhage. Anesth Analg.1999;;89:962–966. [PubMed]
19. Bulsara KR, McGirt MJ, Villavicencio AT, et al. Use of the peak troponin value to differentiate myocardial infarction from reversible neurogenic left ventricular dysfunction associated with aneurismal subarachnoid hemorrhage. J Neurosurg. 2003;;98:524–528. [PubMed]
20. Horowitz MB, Willet D, Keffer J. The use of cardiac troponin I to determine the incidence of myocardial ischemia and injury in patients with aneurysmal and presumed aneurysmal subarachnoid hemorrhage. Acta Neurochir. 1998;;140:87–93.
21. Andreoli A, di Pasquale G, Pinelli G, et al. Subarachnoid hemorrhage: frequency and severity of cardiac arrhythmias. Stroke. 1987;;18:558–564. [PubMed]
22. Brouwers PJ, Wijdicks EF, Hasan D, et al. Serial electrocardiographic recording in aneurysmal subarachnoid hemorrhage. Stroke. 1989;;20:1162–1167. [PubMed]
23. Davies KR, Gelb AW, Manninen PH, et al. Cardiac function in aneurysmal subarachnoid hemorrhage: a study of electrocardiographic and echocardiographic abnormalities. Br J Anaesth.1991;;67:58–63. [PubMed]
24. Kono T, Morita H, Kuroiwa T, et al. Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: neurogenic stunned myocardium. J Am Coll Cardiol. 1994;;24:636–639.[PubMed]
25. Marion DW, Segal R, Thompson ME. Subarachnoid hemorrhage and the heart. Neurosurgery.1986;;18:101–106. [PubMed]
26. Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg. 1995;;83:889–896.[PubMed]
27. Mayer SA, Sherman D, Fink ME, et al. Non-invasive monitoring of cardiac output by Doppler echocardiography in patients treated with volume expansion after subarachnoid hemorrhage.Critical Care Med. 1995;;23:1470–1474. [PubMed]
28. Mayer SA, Lin J, Homma S, et al. Myocardial injury and left ventricular performance after subarachnoid hemorrhage. Stroke. 1999;;30:780–786. [PubMed]
29. Pollick C, Cujec B, Parker S, et al. Left ventricular wall motion abnormalities in subarachnoid hemorrhage: an echocardiographic study. J Am Coll Cardiol. 1988;;12:600–605. [PubMed]
30. Raymer K, Choi P. Concurrent subarachnoid hemorrhage and myocardial injury. Can J Anaesth.1997;;44:515–519. [PubMed]
31. Sakamoto H, Nishimura H, Imataka K, et al. Abnormal Q wave, ST segment elevation, T-wave inversion, and widespread focal myocytolysis associated with subarachnoid hemorrhage. Jpn Circ J.1996;;60:254–257. [PubMed]
32. Sakka SG, Hurettemann E, Reinhart K. Acute left ventricular dysfunction and subarachnoid hemorrhage. J Neurosurg Anesthesiol. 1999;;11:209–213. [PubMed]
33. Solenski N, Haley EC, Kassell NF, et al. Medical complications of aneurysmal subarachnoid hemorrhage: a report of the Multicenter Cooperative Aneurysm Study. Crit Care Med.1995;;23:1007–1017. [PubMed]
34. Zaroff JG, Rordorf GA, Ogilvy CS, et al. Regional patterns of left ventricular systolic dysfunction after subarachnoid hemorrhage: evidence for neurally mediated cardiac injury. J Am Soc Echocardiogr. 2000;;13:774–779. [PubMed]
35. Handlin LR, Kindred LH, Beauchamp GD, et al. Reversible left ventricular dysfunction after subarachnoid hemorrhage. Am Heart J. 1993;;126:235–240. [PubMed]
36. Sato K, Masuda T, Kikuno T, et al. Left ventricular asynergy and myocardial necrosis accompanied by subarachnoid hemorrhage: contribution of neurological pulmonary edema. J Cardiol. 1990;;20:359–367. [PubMed]
37. Diringer MN, Ladenson PW, Stern BJ, et al. Plasma atrial natriuretic factor and subarachnoid hemorrhage. Stroke. 1988;;19:1119–1124. [PubMed]
38. Diringer MN, Lim JS, Hanley DF. Suprasellar and intraventricular blood predict elevated plasma atrial natriuretic factor in subarachnoid hemorrhage. Stroke. 1991;;22:577–581. [PubMed]
39. Mayer SA, Fink ME, Homma S, et al. Cardiac injury associated with neurogenic pulmonary edema following subarachnoid hemorrhage. Neurology. 1994;;44:815–820. [PubMed]
40. Matsuyama N, Masuda T, Yamamoto S, et al. Left ventricular asynergy induced by elevated activity of the noradrenergic nervous system: a study of 717 patients in the acute phase of subarachnoid hemorrhage. Kitasato Med. 1998;;28:494–506.
41. Minegishi A, Ishizaki T, Yoshida Y, et al. Plasma monoaminergic metabolites and catecholamines in subarachnoid hemorrhage: clinical implications. Arch Neurol. 1987;;44:423–428. [PubMed]
42. Szabo MD, Crosby G, Hurford WE, et al. Myocardial perfusion following acute subarachnoid hemorrhage in patients with an abnormal electrocardiogram. Anesth Analg. 1993;;76:253–258.[PubMed]
43. Zaroff JG, Rordorf GA, Titus JS, et al. Regional myocardial perfusion after experimental subarachnoid hemorrhage. Stroke. 2000;;31:1136–1143. [PubMed]
44. Bederson J, Connolly S, Batjer H, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage. Stroke. 2009;;40:994. [PubMed]
45. Chimowitz MI, Mancini GB. Asymptomatic coronary artery disease in patients with stroke: prevalence, prognosis, diagnosis, and treatment. Stroke. 1992;;23:433–436. [PubMed]
46. Myers MG, Norris JW, Hachinski VC, et al. Cardiac sequelae of acute stroke. Stroke.1982;;13:838–842. [PubMed]
47. James P, Ellis CJ, Whitlock RM, et al. Relation between troponin T concentration and mortality in patients presenting with an acute stroke: observational study. BMJ. 2000;;320:1502–1504.[PMC free article] [PubMed]
48. Barasch E, Kaushik V, Gupta R, et al. Elevated cardiac troponin levels do not predict adverse outcomes in hospitalized patients without clinical manifestations of acute coronary syndromes.Cardiology. 2000;;93:1–6. [PubMed]
49. The NINDS and Stroke rtPA Study Group. Tissue plasminogen activator for ischemic stroke. N Engl J Med. 1995;;333:1581–1587. [PubMed]
50. Korosue K, Ishida K, Matsuoka H, et al. Clinical, hemodynamic, and hemorheological effects of isovolemic hemodilution in acute cerebral infarction. Neurosurgery. 1988;;23:148–153. [PubMed]
51. Treib J, Haass A, Koch D, et al. Transcranial Doppler examination on effect of hemodynamics on cerebral autoregulation in acute cerebral infarct. Ultraschall Med. 1996;;17:64–67. [PubMed]
52. Kevorkian GC, Nambiar SV, Rintala DH. Low ejection fraction: effect on the rehabilitation progress and outcome of stroke patients. Am J Phys Med Rehabil. 2005;;84:655–661. [PubMed]
53. Roquer J, Rodríguez-Campello A, Gomis M, et al. Comparison of the impact of atrial fibrillation on the risk of early death after stroke in women versus men. J Neurol. 2006;;253:1484–1489.[PMC free article] [PubMed]
54. Moore C, Rose GA, Tayal VS, et al. Determination of left ventricular function by emergency physician echocardiography of hypotensive patients. Acad Emerg Med. 2002;;9:186–193. [PubMed]
55. Scott PA, Silbergleit R. Misdiagnosis of stroke in tissue plasminogen activator–treated patients: characteristics and outcomes. Ann Emerg Med. 2003;;42:611–618. [PubMed]